How does bisulfite sequencing (WGBS/RRBS) work?
DNA methylation analysis is an increasingly widely-used application of NGS technology. The reason for this is straightforward: DNA methylation is one of the major mechanisms of epigenetic modification, and has a fundamental influence on gene expression and cellular activity (1). This is an interesting aspect for investigating cell development, transcriptional silencing, and biomarker discovery in general.
Why is bisulfite sequencing used?
The aim of NGS-based DNA methylation analysis is to investigate genomic DNA and find out whether single cytosines or entire regions in the genome are methylated or not, since promoter or gene body methylation influences gene expression. Typically, DNA methylation is present in mammals only at CpG dinucleotides, which are methylated by 70 – 85 %, in contrast to CpG islands which are mainly unmethylated to remain active (2). By the way: in humans about 70% of promoters contain a CpG island (3).
There are several methods for studying DNA methylation, but few offer a better resolution of methylation status as bisulfite sequencing (also known as Bisulfite-Seq, BS-Seq or Methyl-Seq). The key idea of this method is combining the power of high-throughput DNA sequencing with the treatment of DNA with sodium bisulfite. When exposed to sodium bisulfite, unmethylated cytosines are converted to uracils whereas methylated forms of cytosine (including 5-Methylcytosine and 5-Hydroxymethylcytosine) remain unchanged (Fig. 1). After sequencing the bisulfite-treated DNA, the obtained sequence reads can be mapped to the original reference genome (Fig. 2) using specialised alignment software which interprets converted bases as unmethylated cytosines instead of errors. The alignment can then be used to resolve methylation status at single nucleotide-level in a similar manner to detecting DNA variants from NGS data. Hereby it is important to keep in mind that bisulfite sequencing cannot distinguish between 5-Methylcytosine (5mC) and 5-Hydroxymethylcytosine (5hmC) even though their functional impact has been found to be different (4).
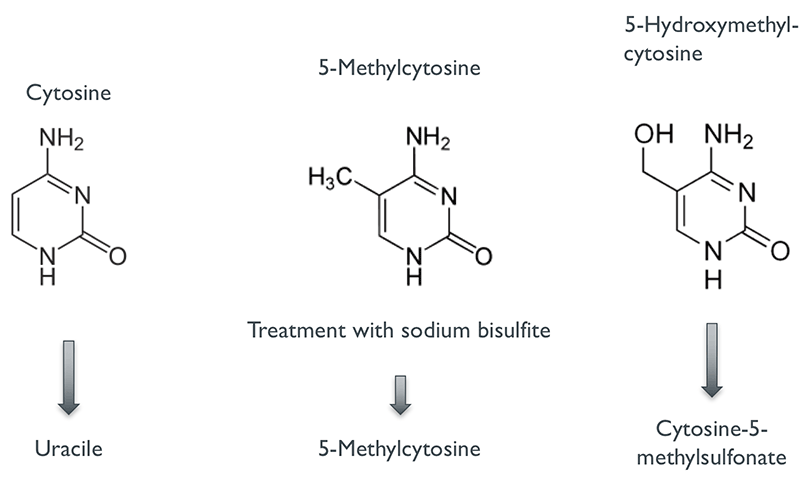 Figure 1: Conversion by bisulfite treatment.
Figure 1: Conversion by bisulfite treatment.
Bisulfite sequencing protocols: Large-scale versus small-scale
There are different protocols that you can use to assess DNA methylation using NGS. The easiest way surely is to add the bisulfite reaction to your sequencing workflow and do Whole-Genome Bisulfite Sequencing (WGBS). However, you will need sufficient read depths to reliably determine methylation status. When you are working on an organism with a large genome size, this can lead to high costs for sequencing.
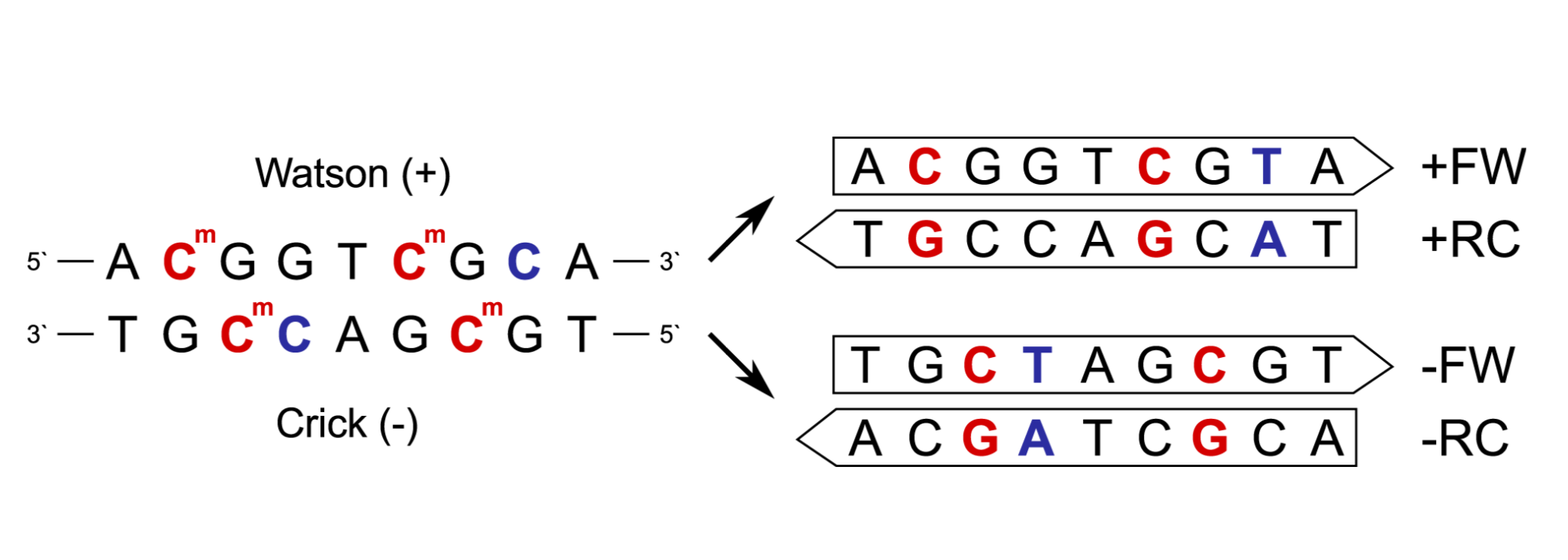 Figure 2: Read types after sequencing.
Figure 2: Read types after sequencing.
As an alternative, you could focus the detection of DNA methylation to a specific subset of the genome, thereby reducing the data volume of your experiment and subsequently the cost.
One popular approach to this is Reduced Representation Bisulfite Sequencing (RRBS). The fundamental idea of RRBS is to get a “reduced representation" of the genome, with a focus on CpG islands. This involves the addition of restriction enzymes to digest the DNA during the fragmentation step. Typically, the enzyme MspI is used which is methylation insensitive. It cuts at 5’-CCGG-3’ sites, and since the genome is largely depleted of CpGs except for promoters/CpG islands, the "reduced representation" is largely capturing only these promoter regions for further analysis. The digestion reaction enriches for DNA fragments with a CpG at each end and fragments of various sizes. The fragment ends are then filled in and adapters are ligated. Afterwards the fragments are size selected, bisulfite-converted, and sequenced.
Concluding remarks
One last piece of advice for bisulfite sequencing in general: since the treatment of DNA with bisulfite basically destroys your DNA, it is important to check whether the remaining DNA has intact adapters at both ends before sequencing with qPCR or other techniques. Otherwise, one might end up with a sequencing library of low complexity.
In addition to WGBS and RRBS, there are enrichment kits available for human DNA that represent a cost-effective solution for analysing promoter and gene body methylation. It is also possible to get custom methylation enrichment kits for your regions of interest and your organism.
We hope this article was helpful for understanding how bisulfite sequencing works and how it differs between WGBS and RRBS in general use cases.
References:
- Jones, P. A., & Takai, D. The role of DNA methylation in mammalian epigenetics. Science, 293(5532), 1068-1070 (2001).
- Jabbari, K. & Bernardi, G. Cytosine methylation and CpG, TpG (CpA) and TpA frequencies. Gene 333, 143–149 (2004).
- Saxonov, S., Berg, P. & Brutlag, D. L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. U. S. A. 103, 1412–1417 (2006).
- Branco, M. R., Ficz, G. & Reik, W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nature Reviews Genetics. 13(1), 7 (2012).
Would you like to sharpen your NGS data analysis skills?
Join one of our public workshops!About us
ecSeq is a bioinformatics solution provider with solid expertise in the analysis of high-throughput sequencing data. We can help you to get the most out of your sequencing experiments by developing data analysis strategies and expert consulting. We organize public workshops and conduct on-site trainings on NGS data analysis.
Last updated on September 17, 2018