Are there regions in the genome that are not covered by DNA sequencing?
If the genome would be a landscape, and you would create a map of that landscape by NGS, the map would certainly have some white areas. In other words: some genomic regions cannot be covered very well by sequencing the DNA with NGS technology. Here, the most important reasons for this will be explained.
Repetitive DNA sequences
Repetitive DNA sequences (e. g. Tandem repeats) are abundant in a broad range of species. Approximately 50% of the human genome is comprised of repeats (1). Repeats are a challenge for sequence alignment and assembly programs because they lead to very similar short reads. It can be compared to a big puzzle whereby some pieces of the puzzle fit at several places. Thus repeats create ambiguities in alignment and assembly, which, in turn, can produce biases and errors when interpreting results (1).
G/C Bias
The four bases (ACTG) are normally not equally distributed in a genome. DNA regions with high and low GC contents are hard to amplify because of a higher stability compared to a DNA region with a mixed base content. In these cases, DNA polymerases is prone to produce artefacts. These effects disturb the amplification steps required in most protocols. Consequently fragments from regions with high/low GC content are underrepresented, resulting in a poor and unbalanced read coverage (Fig. 1).
A common example for this is Plasmodium falciparum, a malaria pathogen. Some of the coding regions of its genome have 70% AT content. Thus, until recently, it was not possible to sequence the parasite's genome. Current projects try to improve sequencing of such regions by either minimizing the artefacts produced by PCR (amplification) or renouncing the amplification step (2).
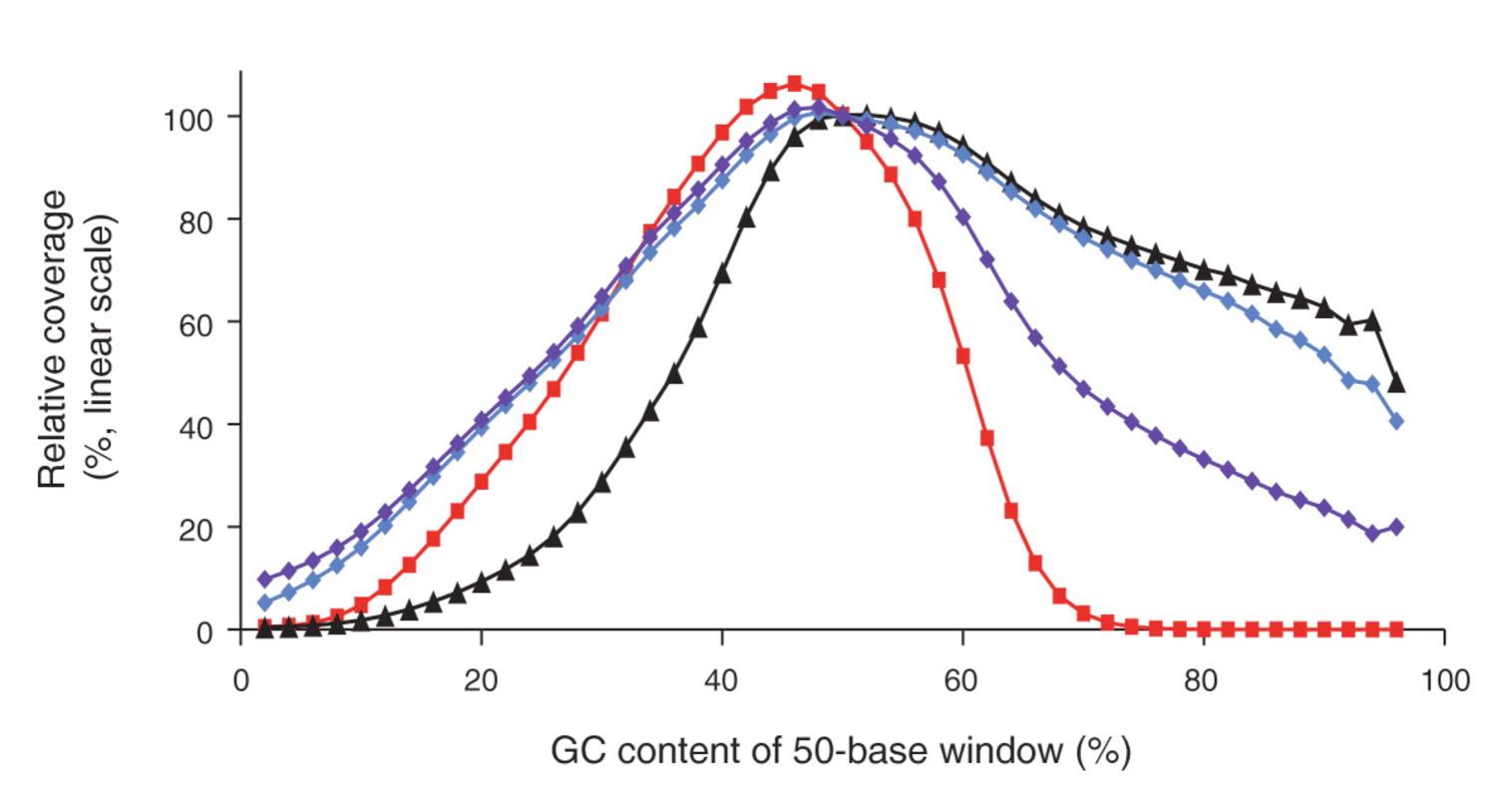 Figure 1: Genome-wide base composition bias curves under various amplification conditions, linear scale. Image from (3).
Figure 1: Genome-wide base composition bias curves under various amplification conditions, linear scale. Image from (3).
Fragmentation Bias
As described in this article, DNA fragmentation is a largely non-random process, especially for mechanical shearing methods. This leads to a non-uniform coverage of various genomic regions and can result in under/uncovered regions.
Summary
These examples show that there are quite some technical challenges yet to be solved for accurate DNA sequencing using NGS. Sometimes it is possible to reduce the issues by adapting sample preparation (e. g. for DNA fragmentation) or the sequencing process itself (as described for Plasmodium falciparum). Additionally it is always a good idea to consider different sequencing technologies for a project.
- Treangen, T. J. & Salzberg, S. L. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 13, 36–46 (2011).
- Gardner, M. J. et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419, 498–511 (2002).
- Aird, D. et al. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 12, R18 (2011).
Would you like to sharpen your NGS data analysis skills?
Join one of our public workshops!About us
ecSeq is a bioinformatics solution provider with solid expertise in the analysis of high-throughput sequencing data. We can help you to get the most out of your sequencing experiments by developing data analysis strategies and expert consulting. We organize public workshops and conduct on-site trainings on NGS data analysis.
Last updated on May 06, 2017