What is mate pair sequencing for?
You certainly have heard of mate pair sequencing. It’s a smart technique that allows you to obtain paired-end reads with long inserts. That makes it a powerful tool for various sequencing applications including de-novo genome sequencing. Here we will discuss how it works and what are its applications in detail.
Overview
To simplify, you can differ between two kinds of reads for paired-end sequencing: short‑insert paired‑end reads (SIPERs) and long-insert paired-end reads (LIPERs). The latter one is also called mate pair. The difference between the two variants is first – surprise - the length of the insert. SIPERs are 200‑800 bp long, LIPERs can be longer.
Definitely more interesting is the difference in the way the two are created. Making SIPERs is not very spectacular: after fragmenting genomic DNA you can isolate fragments of your desired length (200-800 bp) and ligate adapters to them (Fig. 1). If you want to use longer inserts to cover a larger distance between the reads you have a problem: it is not feasible to use insert sizes over 1 kb.
Luckily this is not the end of the story. There is a nice trick used for mate pair sequencing, shown in Fig. 1. First DNA is fragmented and fragments of a desired length (2-5 kb) are isolated. Afterwards the ends of the DNA fragments are biotinylated (adding Biotine). The biotinylated ends leads to a circularizing of the fragments. Then the DNA ring is crushed into smaller fragments (400-600 bp). Biotinylated fragments are enriched (by biotin tag) and adapters are ligated. They are then ready for cluster generation and sequencing. The trick here is that the produced fragment (400-600 bp) contains the ends of the original long fragment (2-5 kb) and can be sequenced now. After sequencing you therefore get information about the original fragment.
 Figure 1: Comparison of sample preparation for Illumina Paired-End Sequencing and Illumina Mate Pair Sequencing
Figure 1: Comparison of sample preparation for Illumina Paired-End Sequencing and Illumina Mate Pair Sequencing
Application of mate pair sequencing
Mate pair sequencing is used for various applications applications, including
- De novo genome sequencing
- Genome finishing
- Structural variant detection
- Identification of complex genomic rearrangements
Combining data from mate pair sequencing with those from short-insert paired-end reads provides increased information for maximising sequencing coverage across a genome (1). This can be very helpful, e. g. for your De novo genome assembly (Fig. 2). The larger inserts (mate pairs) can pair reads across greater distances. Therefore they are able to better cover highly repetitive regions. Short-insert paired-end reads can fill in gaps missed by larger mate pairs (Fig. 2). This combination leads to larger contigs and greater accuracy of the final consensus sequence (1).
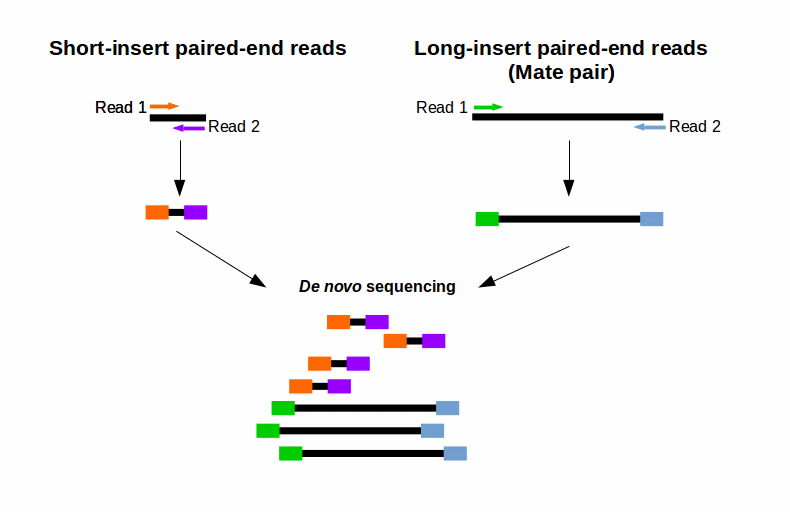 Figure 2: Combining reads from mate pair sequencing with that from short-insert paired-end sequencing for De-novo Sequencing.
Figure 2: Combining reads from mate pair sequencing with that from short-insert paired-end sequencing for De-novo Sequencing.
Conclusion
As you can see mate pair sequencing is a helpful sequencing technique which helps to reduce the problems resulting from the length limitations of SIPERs. Moreover mate pair and SIPERs can be combined in efficient ways. The applications in the list above are just recommendations and certainly there are many more possible uses so try and find out.
Would you like to sharpen your NGS data analysis skills?
Join one of our public workshops!About us
ecSeq is a bioinformatics solution provider with solid expertise in the analysis of high-throughput sequencing data. We can help you to get the most out of your sequencing experiments by developing data analysis strategies and expert consulting. We organize public workshops and conduct on-site trainings on NGS data analysis.
Last updated on March 20, 2017