Why has the reverse read 2 a worse quality than the forward read 1 in Illumina sequencing?
It is well known that in Illumina sequencing, the per base quality decreases towards the end of the sequence reads. Why that happens is explained here. But when you work with paired-end sequencing, you will often notice that read 2 (the reverse read) has a worse quality than read 1. More precisely, the base quality decreases much earlier towards the end of the reverse read compared to the the forward read.
When comparing the two FASTQC image below, the effect will directly catch your eye. What is the reason for this phenomenon and is there a way to improve the reverse read quality?
 Figure 1: Per base sequence quality plot for forward and reverse read, respectively.
Figure 1: Per base sequence quality plot for forward and reverse read, respectively.
The amplification problem
Let’s start with the good news: the worse quality of read 2 is probably not your fault. A lower quality for the reverse read is an expected phenotype in paired-end sequencing runs. This is explained by the fact that the clusters size decreases during bridge amplification at the paired-end turnaround stage (see Figure 2) that occurs before read 2 is sequenced. During the paired-end turnaround, an Illumina MiSeq does 12 cycles of bridge amplification in order to regenerate the clusters.
Both, 1) a cluster with a smaller amount of molecules and 2) a higher number of errors within these molecules due to more amplification steps, lead to the effect that the per base quality of the read 2 cluster decreases much earlier than for read 1. The explanation: The already increased percentage error rate within the (smaller) cluster is now added to the normal 'phasing errors'.
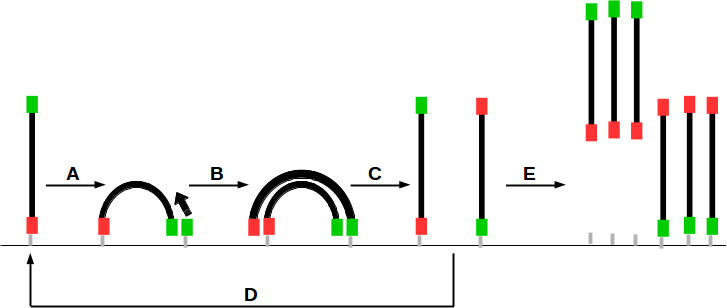 Figure 2: Paired-end turnaround stage after sequencing read 1 resulting in growing clusters. A: Bridge formation (hybridisation), B: Bridge amplification, C: Bridge dissolve, D: Return to A (12 cycles for MiSeq), E: Replacing of P7-fixed fragments. Flow cell binding sites: P5 (green) and P7 (red).
Figure 2: Paired-end turnaround stage after sequencing read 1 resulting in growing clusters. A: Bridge formation (hybridisation), B: Bridge amplification, C: Bridge dissolve, D: Return to A (12 cycles for MiSeq), E: Replacing of P7-fixed fragments. Flow cell binding sites: P5 (green) and P7 (red).
How to avoid the worse quality of read 2?
In summary, the reverse read quality problem is not really solvable, since a higher number of amplification steps for cluster regeneration after paired-end turnaround will result in a cluster consiting of more molecules, but also in a higher number of errors between the molecules within this cluster. Following the guidlines from Illumina will likely result in an optimized ratio between errors and cluster size.
Would you like to sharpen your NGS data analysis skills?
Join one of our public workshops!About us
ecSeq is a bioinformatics solution provider with solid expertise in the analysis of high-throughput sequencing data. We can help you to get the most out of your sequencing experiments by developing data analysis strategies and expert consulting. We organize public workshops and conduct on-site trainings on NGS data analysis.
Last updated on February 17, 2020