Blog News from ecSeq
Meet Martin Kircher - Ancient DNA and Genomics Expert at the Berlin Summer School in NGS Data Analysis
Posted on February 04, 2026

We are very pleased to announce that Prof. Dr. Martin Kircher will be joining us as an invited speaker at the upcoming Berlin Summer School in NGS Data Analysis this July.
Martin is a leader in the fields of computational genomics and bioinformatics. Since 2017, he heads the Computational Genome Biology research group at the Berlin Institute of Health (BIH) at Charité – Universitätsmedizin Berlin, and in 2022 he was appointed Professor of Regulatory Genomics at the University of Lübeck and the University Hospital Schleswig-Holstein.
His research focuses on computer-based methods to identify functionally relevant genomic sequences and sequence variants, and on developing sensitive analyses for sequencing data, including exome, whole genome, and cell-free DNA sequencing. His team also develops widely used variant effect scoring tools such as CADD, CADD-SV and ReMM, and integrates experimental data from high-throughput assays such as reporter screens and CRISPR perturbations into predictive models of regulatory sequence function.
Martin’s work reflects a deep commitment to advancing how we analyse both coding and non-coding genomic variation, applying machine learning, data mining and functional genomics to understand genetic effects in disease and evolution. His expertise bridges methodological innovation and practical bioinformatics challenges — a connection that aligns closely with the spirit of hands-on learning we aim to foster at the Summer School.
At the Summer School, Martin will share insights from his research and discuss practical challenges in analysing complex sequencing datasets. His talk will provide participants with a view of how cutting-edge genomic analysis tools are developed and applied in real research contexts, especially in the challenging domain of variant effect prediction and functional genome interpretation.
The Berlin Summer School in NGS Data Analysis is designed to give attendees a comprehensive, hands-on experience with next-generation sequencing workflows — from initial quality control and mapping to advanced topics such as variant interpretation, single-cell analysis, and reproducible pipeline development.
We are very much looking forward to welcoming Martin to Berlin and hearing his perspective on computational genomics.
For more details and to apply, please visit the Summer School page:
ecseq.com/summer-school
Registration is now open — we encourage you to apply early as spaces are limited.

On-site Training in Zagreb: A Success Story with Sanja Duvnjak and the Croatian Veterinary Institute
Posted on February 02, 2026

We are delighted to share a highlight from the end of 2025, when ecSeq Bioinformatics delivered tailored on-site NGS and bioinformatics training at the Croatian Veterinary Institute in Zagreb. This engagement grew out of an earlier connection with Sanja Duvnjak, a long-standing participant in our workshops, who found our introductory course so transformative that she invited us to bring the training directly to her team in Croatia.
For context, Dr. Sanja Duvnjak is Head of the Laboratory for Bacterial Zoonoses and Molecular Diagnostics of Bacterial Diseases at the Croatian Veterinary Institute in Zagreb, a key research centre in veterinary and zoonotic disease analytics with decades of scientific excellence. Her work encompasses microbial molecular biology, zoonoses diagnostics, and genomic approaches to understanding bacterial pathogens, reflecting a deep commitment to applied science and practical training.
We were warmly welcomed to Zagreb, where we conducted a 3-day introductory course followed by a 2-day Nextflow pipeline workshop. The introductory module brought together ten participants, helping them build foundational competence in NGS data analysis — from understanding how sequencing data are generated to implementing their first analysis pipelines with confidence. In the advanced Nextflow module, four participants learned how to turn these pipelines into robust, reproducible workflows that now serve as internal, reusable infrastructure.
Here is Sanja’s own account of the training experience:
"At the end of 2025, ecSeq Bioinformatics delivered a tailored on-site NGS and bioinformatics training at the Croatian Veterinary Institute in Zagreb. The course was carefully designed to meet both our immediate analytical needs regarding basic bioinformatics analysis and our long-term strategic goals for pipeline development. In the introductory module, ten participants learned how NGS data are generated, which scientific questions can be addressed with sequencing technologies, and how bioinformatics workflows transform raw data into interpretable results. Participants then implemented their first analysis pipeline in a guided, step-by-step manner. The individual tools and intermediate results were explained in depth, enabling participants to critically assess and interpret the output rather than treating the pipeline as a black box. In the advanced module, five participants learned how to put this pipeline into a robust, reproducible, and automated workflow using Nextflow. This has empowered us to develop a reusable internal pipeline for routine data analysis and future projects. The course was very informative and allowed for future collaborations and data interpretation to non-bioinformatic staff who are going to use the analysis outputs. The on-site training was time and money-saving, providing education to staff that wouldn't otherwise get that opportunity in a manner that was understandable even to those who were dealing with this kind of analysis for the first time. The communication and the organisation were very simple and easy-going, and most importantly, what was promised and expected was delivered. We hope to continue this collaboration in the future."

This glowing testimonial reflects exactly what we aim to achieve with our on-site training programmes: practical, impactful education delivered where teams work and where it matters most.
ecSeq’s on-site courses are designed to be flexible, team-focused, and aligned with your organisation’s specific needs. Whether an introductory three-day course, targeted advanced modules, or pipeline automation training with tools like Nextflow, our experts work side by side with your team to build confidence, skills, and reproducible workflows. Find out more about our on-site offerings here.
We are extremely grateful to Sanja and the Croatian Veterinary Institute for their hospitality, enthusiasm, and collaborative spirit. Building capacity together with committed teams like theirs is one of the most rewarding aspects of what we do, and we look forward to more such collaborations in the future.
If your team is interested in tailored on-site bioinformatics and NGS training, we’d be delighted to speak with you about how we can design a programme that fits your goals.

Meet Vladimir Jovanovic - Bioinformatics Expert at the 10th Berlin Summer School in NGS Data Analysis
Posted on February 02, 2026

We are excited to welcome Dr. Vladimir Jovanović from the Freie Universität Berlin as an invited speaker at the upcoming 10th Berlin Summer School in NGS Data Analysis (May 15–19, 2026). Vladimir will contribute his expertise and unique perspective to the programme, enriching the learning experience for all participants.
Vladimir is a postdoctoral researcher affiliated with the Bioinformatics Solution Center and the Nowick Group – Human Biology at Freie Universität Berlin, where he works at the interface of bioinformatics, genomics, and evolutionary biology. His research interests span the evolution and specific functions of gene regulatory factors in primates, investigations of random monoallelic expression in neurodegenerative diseases such as Alzheimer’s, and broader questions in human genetic disease and population variation. His interdisciplinary background gives him a deep appreciation for both the biological questions and the computational challenges that arise in modern sequencing projects.
In addition to his academic research, Vladimir has contributed bioinformatic analysis expertise to a range of collaborative projects. For example, he was involved in recent work that provided novel insights into genetic relatedness structures in free-ranging primate populations — illustrating how careful computational analysis can reveal patterns of inheritance that traditional pedigree approaches might miss.
At the Summer School, Vladimir will share insights from his research and work that highlight practical aspects of bioinformatics data analysis, bridging the gap between biological complexity and computational methodology. His contribution will complement the core curriculum by offering real-world examples and discussion points that reflect current challenges in NGS and genomic research.
The 10th Berlin Summer School in NGS Data Analysis offers an intensive, hands-on introduction to the key tasks and tools of next-generation sequencing, including quality control, mapping, variant calling, and downstream interpretation. The programme is designed for those new to NGS bioinformatics as well as learners looking to solidify their understanding of practical workflows.
We are very much looking forward to Vladimir’s session and are delighted that he will be joining our community in Berlin this May.
For more details and to apply, please visit the Summer School page: www.ecseq.com/summer-school
Applications are open now — we encourage you to apply early as spaces are limited.
A Practical Introduction to NGS Data Analysis and Variant Calling, January 2026 (Online)
Posted on January 30, 2026

A truly international NGS workshop: Our January 2026 online introduction
At the end of January, we ran our online “A Practical Introduction to NGS Data Analysis and Variant Calling” workshop, and it turned out to be a truly international and very rewarding experience for everyone involved.
Over three intensive days, we welcomed participants from France, Slovenia, Saudi Arabia, Algeria, Greece, Poland, New Caledonia, Australia, the United Kingdom, Germany, Bulgaria, and Austria. Having such a geographically diverse group always adds something special to the course — different backgrounds, different research questions, and a wide range of perspectives that enrich the discussions.
From the very first session, the atmosphere was excellent. Participants asked many thoughtful and highly relevant questions, leading to lively exchanges and deep dives into both practical details and conceptual aspects of NGS data analysis. These questions are always one of the highlights for us as trainers, as they reflect real challenges people face when working with sequencing data in their own projects.
The three days were undoubtedly intense and demanding — online workshops require sustained focus from everyone involved. Nevertheless, we managed to work through the complete program as planned, and we hope that all participants were able to follow the material well and leave the course with a clearer understanding and greater confidence in their skills.
We very much enjoyed spending these three days together with such an engaged and motivated group, and we are grateful for the open and constructive learning atmosphere that developed despite the virtual setting. We sincerely hope that we will meet many of the participants again in the future — perhaps in another online course, an in-person workshop, or even at one of our on-site trainings.
We regularly offer introductory and advanced NGS data analysis courses, both online and in person, and we are always happy to welcome new participants from around the world.
Thank you again to everyone who joined us in January — it was a pleasure to learn and work together.
Meet Jeremy Leipzig - TileDB Expert at the 10th Berlin Summer School in NGS Data Analysis
Posted on January 29, 2026
We are delighted to announce that Jeremy Leipzig, Senior Product Manager for Genomics at TileDB, will be joining us as an invited speaker at the upcoming 10th Berlin Summer School in NGS Data Analysis (May 15–19, 2026). Jeremy will deliver a short session introducing TileDB-VCF, a scalable and flexible approach to working with genomic variant data.
Jeremy brings extensive experience in bioinformatics and data engineering to the Summer School. He holds a PhD and has worked across academia, industry, and research settings focused on genomic data, reproducibility, workflows, and high-volume variant storage. Before joining TileDB, Jeremy contributed to projects in virology, crop genomics, and pediatric research, often emphasizing the importance of reproducible analysis and metadata management — topics that resonate strongly with modern NGS projects.
At TileDB, he focuses on unlocking scalable and reproducible data solutions in genomics. In particular, TileDB-VCF provides an alternative to traditional VCF file handling by storing and querying variant data in a highly efficient database format. This enables researchers and bioinformaticians to work interactively with large cohorts, perform federated queries, and manage multi-sample variant data without the typical bottlenecks of file-based workflows.
Participants at the Berlin Summer School will have the unique opportunity to gain insight into how TileDB-VCF and related technologies can support real-world genomics projects, especially those involving large cohorts or complex variant datasets. Jeremy's presentation will complement our core curriculum by combining practical NGS workflows with new innovations in data infrastructure.
The 10th Berlin Summer School in NGS Data Analysis is designed to give attendees a comprehensive, hands-on experience in next-generation sequencing workflows, from initial quality control and mapping to advanced topics such as variant calling, single-cell analysis, and reproducible workflow development. The addition of expert contributions like Jeremy’s ensures that the programme remains at the cutting edge of both analysis practice and data management strategy.
We are very much looking forward to welcoming Jeremy to Berlin and to hearing his insights on scalable genomic data approaches.
For more details and to apply, please visit the Summer School page:
ecseq.com/summer-school
Applications are open now — we encourage you to apply early as spaces are limited.
NGS Epigenomics Workshop, November 2025
Posted on November 05, 2025

The recent NGS Epigenomics with ChIP-Seq and Bisulfite-Seq Workshop, held in Nomveber 2025, provided participants with a deep and practical introduction to modern epigenomic analysis workflows.
A total of 10 researchers took part, coming from eight countries. The strong international character of the course was clearly visible, with three attendees from Germany, and additional participants joining from Saudi Arabia, Poland, Italy, the United States of America, the Netherlands (2), and New Zealand.
Over the course of the workshop, participants gained hands-on experience with state-of-the-art tools for analyzing ChIP-Seq and bisulfite sequencing data. These practical sessions were designed to strengthen technical skills and empower attendees to confidently apply epigenomic methods in their own research projects.
This intensive training format continues to support scientists in bridging knowledge gaps in Next-Generation Sequencing (NGS)-based epigenomics, equipping them with essential expertise for successful independent and collaborative bioinformatics work. For insights into the participants' experiences and their feedback on the course, please visit here.
Single-Cell RNA-Seq Data Analysis Workshop, October 2025
Posted on October 13, 2025

The recent Single-Cell RNA-Seq Data Analysis Workshop, held in Berlin from October 6 to 8, 2025, offered a comprehensive immersion into single-cell RNA-Seq data analysis.
The event successfully attracted 19 participants from six European nations. The strong international interest was evident, with the majority of attendees from Germany (15), and additional groups from Italy (2) and Finland (2), as well as single participants arriving from Norway, Slovakia, and Denmark.
The core of the workshop involved intensive hands-on sessions, during which participants employed advanced tools to analyze single-cell RNA-Seq data. This practical focus was designed to significantly enhance their technical capabilities for both independent and collaborative genomic research.
Ultimately, the course aims to bridge the knowledge gap in Next-Generation Sequencing (NGS) data analysis, providing researchers with the essential tools and confidence to confidently advance their bioinformatics projects. For insights into the participants' experiences and their feedback on the course, please visit here.
9th Berlin Summer School in NGS Data Analysis 2025
Posted on July 07, 2025

The 9th Berlin Summer School in NGS Data Analysis (June 30 – July 4, 2025) was a fantastic success — five days packed with cutting-edge learning, international networking, and hands-on training. We welcomed participants from around the world, creating a vibrant and highly interactive learning environment. From day one, the energy was inspiring: insightful questions, lively discussions, and plenty of laughter made this an unforgettable experience for both attendees and trainers.
A special highlight of the week was the inclusion of two invited talks from leading researchers in the NGS field. Their real-world insights into current applications and breakthroughs added exceptional value and sparked enthusiastic discussions.
Designed to empower researchers with essential skills in NGS data analysis, this course is the perfect launchpad into the world of bioinformatics. Whether you're new to the field or looking to strengthen your foundation, our training combines practical knowledge with expert guidance.
Curious about what our participants had to say? Check out their feedback and impressions here.
Introduction to NGS Data Analysis Workshop, April 2025 in Munich
Posted on April 07, 2025

Our recent Introduction to NGS Data Analysis Workshop in Munich, held from March 12 to 14, 2025, brought together an exceptional group of 16 participants from Hong Kong, Austria, Germany, Poland, the Netherlands, Spain, Italy, and the United Kingdom.
Throughout the three days, the workshop was marked by lively discussions and a shared curiosity that energized the learning environment. Participants asked thoughtful and challenging questions, sparking valuable insights for both their peers and the trainers. This open exchange fostered a collaborative spirit that made the workshop both enriching and enjoyable.
Hands-on sessions focused on essential NGS data analysis techniques, including command-line operations, quality control, and the interpretation of sequencing data. The diverse backgrounds of the attendees added depth to conversations and enabled fruitful networking opportunities.
We’re confident this experience will empower participants to take on their future data analysis projects with clarity and confidence. If you’d like to be part of a similarly inspiring learning environment, explore our upcoming workshops—we’d love to welcome you!
RNA-Seq Data Analysis Workshop, March 2025
Posted on April 01, 2025

The recent RNA-Seq Data Analysis Workshop, held in Halle from March 24–27, 2025, offered a deep dive into the practical aspects of bulk RNA-Seq data analysis. This intensive four-day workshop brought together 15 participants from Belgium, Germany, and the Czech Republic, creating a dynamic and collaborative learning environment.
With a strong focus on hands-on training, the course guided participants through each step of a standard RNA-Seq analysis workflow—from quality control and read alignment to differential expression and result interpretation. The interactive sessions were supported by expert trainers, who provided both technical guidance and valuable insights into real-world applications of RNA-Seq in modern life science research.
Whether new to RNA-Seq or looking to strengthen existing skills, attendees left the workshop equipped with the tools and confidence to tackle their own datasets effectively.
The course is designed to bridge the knowledge gap in NGS data analysis, providing researchers with the tools and confidence needed to embark on their bioinformatics journey. For insights into the participants' experiences and their feedback on the course, please visit here.
Single-Cell RNA-Seq Data Analysis Workshop, March 2025
Posted on March 24, 2025

The recent Single-Cell RNA-Seq Data Analysis Workshop in Berlin, spanning March 17 - 19, 2025, , provided a comprehensive learning experience in single-cell RNA-Seq data analysis. The event drew 22 participants from countries including Germany, Poland, Denmark, Israel, Ireland, Czech Republic, United Kingdom and Italy, underscoring its global appeal. The workshop featured intensive hands-on sessions where participants used advanced tools to analyze single-cell RNA-Seq data, enhancing their skills for both collaborative and independent genomic research.
The course is designed to bridge the knowledge gap in NGS data analysis, providing researchers with the tools and confidence needed to embark on their bioinformatics journey. For insights into the participants' experiences and their feedback on the course, please visit here.
We are Moving to Bluesky - Join Us!
Posted on December 18, 2024

A Fresh Start for Open Science
After much consideration, we have decided to leave X (formerly Twitter) and transition to Bluesky, a platform where the scientific community thrives and open, honest discussions about science can flourish.
This decision comes in response to the increasing hostility on X, the spread of misinformation, and the distortion of facts driven by its algorithms. The environment has become unwelcoming to scientists, and we believe it’s time to take a stand for free and unbiased science communication.
Bluesky offers a refreshing alternative—a space where the scientific community can engage constructively, without fear of censorship or misinformation. We’re excited to join this growing network and continue sharing our insights with a community that values truth and collaboration.
Follow us on Bluesky: ecseq.bsky.social
Let’s give free science a chance and build a better platform for open communication together.
Pioneering Precision Medicine: ecSeq in SaxoCell® Phase 2
Posted on December 16, 2024
ecSeq Accelerates Cutting-Edge Research in SaxoCell’s Second Phase
For over three years, ecSeq Bioinformatics has been an integral part of SaxoCell—one of Germany’s most prestigious “Clusters4Future” initiatives. As the cluster enters its highly anticipated second phase, we are thrilled to continue contributing to groundbreaking advancements in gene and cell therapeutics, also known as “living drugs.” SaxoCell’s mission to transform precision medicine and revolutionize treatment options for severe diseases aligns perfectly with our expertise in bioinformatics.
Advancing into the Second Phase
The activation of SaxoCell’s second funding phase marks a major milestone in the cluster’s journey. Building on the innovative groundwork laid during the initial phase, this next stage focuses on scaling therapeutic breakthroughs for clinical applications. By integrating interdisciplinary expertise and enhancing technology transfer, the cluster aims to bring advanced therapies closer to patients while positioning Saxony as a global hub for biotechnology.
The Impact of SaxoCell’s Vision
SaxoCell’s second phase is not just about innovation; it’s about making innovation accessible and impactful. With an emphasis on automating production processes and exploring allogeneic therapy approaches, the cluster is working to make gene and cell therapies more affordable and widely available. By leveraging our bioinformatics expertise, we support this vision.
Driving Excellence Together
Being part of SaxoCell is more than a partnership; it’s a commitment to excellence and a shared vision for the future of healthcare. With SaxoCell’s interdisciplinary collaboration, we are advancing precision medicine while reinforcing ecSeq’s position as a leader in bioinformatics. The second phase represents an exciting opportunity to push boundaries, solve complex problems, and contribute to therapies that have the potential to transform countless lives.
As SaxoCell progresses into this critical new chapter, we at ecSeq remain dedicated to supporting its mission with cutting-edge bioinformatics tools and expertise. Together, we are shaping the future of gene and cell therapeutics—making the impossible possible, one discovery at a time.
Single-Cell RNA-Seq Data Analysis Workshop, September 2024
Posted on September 30, 2024

The recent Single-Cell RNA-Seq Data Analysis Workshop in Berlin, held from September 25-27, 2024, offered a comprehensive and immersive experience in analyzing single-cell RNA-Seq data. We were thrilled to welcome 17 participants from countries including Germany, the Czech Republic, Hungary, Austria, Switzerland, and the United States. The hands-on sessions allowed attendees to use advanced bioinformatics tools, significantly boosting their skills for both collaborative and independent research projects.
The course is designed to bridge the knowledge gap in NGS data analysis, providing researchers with the tools and confidence needed to embark on their bioinformatics journey. For insights into the participants' experiences and their feedback on the course, please visit here.
Introduction to NGS Data Analysis Workshop, September 2024 in Munich
Posted on September 05, 2024

Our recent Introduction to NGS Data Analysis Workshop in Munich, held from September 2 to 4, 2024, was a fantastic blend of learning and collaboration. We had the pleasure of welcoming 19 participants from Denmark, the United States, Austria, Ireland, Saudi Arabia, and Germany.
The discussions throughout the workshop were incredibly insightful, with participants asking thought-provoking questions that enriched the learning experience for everyone, including the trainers. The group’s enthusiasm and intelligence fostered dynamic exchanges, which helped not only the attendees but also the trainers to gain new perspectives.
Hands-on sessions allowed participants to dive deep into the practical aspects of NGS data analysis, mastering key skills like command-line navigation and data interpretation. The variety of backgrounds contributed to engaging networking opportunities, adding a valuable professional development layer.
We’re excited to see the impact this workshop will have on their future research. If you're eager to enhance your skills and engage with like-minded professionals, don’t miss our upcoming workshops. Join us in this journey of discovery and learning!
8th Berlin Summer School in NGS Data Analysis 2024
Posted on June 17, 2024

The recent 8th Berlin Summer School in NGS Data Analysis, held from June 10 - 14, 2024, was a remarkable event. With 30 participants from Switzerland, the Czech Republic, Slovenia, Serbia, Germany, Saudi Arabia, Sweden, Austria, Italy, France, the United States, Poland, and the UAE, the workshop underscored its global appeal. The group was highly engaged, asking excellent questions and collaborating effectively, making it a pleasure to work with them. There was a lot of laughter, chatting, and productive discussions.
The workshop also featured two invited talks about research with NGS, which were incredibly insightful and fascinating. These talks added tremendous value to the overall learning experience.
The course is designed to bridge the knowledge gap in NGS data analysis, providing researchers with the tools and confidence needed to embark on their bioinformatics journey. For insights into the participants' experiences and their feedback on the course, please visit here.
Bioinformatics Pipeline Development with Nextflow Workshop, May 2024
Posted on June 07, 2024
The recent Bioinformatics Pipeline Development with Nextflow Workshop, held from May 27 - 29, 2024, offered an immersive experience in developing bioinformatics pipelines. The event attracted 12 participants from countries including Israel, Germany, the USA, the United Kingdom, and Italy, highlighting its international appeal. The workshop featured intensive hands-on sessions where participants used advanced tools to build and optimize bioinformatics pipelines, enhancing their skills for both collaborative and independent research.
The course is designed to bridge the knowledge gap in NGS data analysis, providing researchers with the tools and confidence needed to embark on their bioinformatics journey. For insights into the participants' experiences and their feedback on the course, please visit this page.
Single-Cell RNA-Seq Data Analysis Workshop, May 2024
Posted on May 13, 2024

The recent Single-Cell RNA-Seq Data Analysis Workshop in Berlin, spanning May 6 - 8, 2024, , provided a comprehensive learning experience in single-cell RNA-Seq data analysis. The event drew 14 participants from countries including Germany, Poland, Spain, the Czech Republic, Switzerland, and Canada, underscoring its global appeal. The workshop featured intensive hands-on sessions where participants used advanced tools to analyze single-cell RNA-Seq data, enhancing their skills for both collaborative and independent genomic research.
The course is designed to bridge the knowledge gap in NGS data analysis, providing researchers with the tools and confidence needed to embark on their bioinformatics journey. For insights into the participants' experiences and their feedback on the course, please visit here.
Exploring the Power of Single-Cell RNA Sequencing in Modern Research
Posted on April 22, 2024
Single-cell RNA sequencing (scRNA-seq) represents a revolutionary approach in genomics, enabling scientists to examine the transcriptomic details at the individual cell level. This powerful technique offers a window into the complexity of biological systems, uncovering the diverse roles of single cells in health and disease.
What is Single-Cell RNA Sequencing?
scRNA-seq is a method used to profile the gene expression of individual cells. By isolating single cells and sequencing their RNA, researchers can identify which genes are active in each cell at a given moment. This level of detail is crucial for understanding cellular diversity and function within a mixed population of cells.
Capabilities of Single-Cell RNA Sequencing:
- Cellular Diversity: scRNA-seq reveals the existence of different cell types within a tissue, even those that are rare or previously undefined.
- Disease Mechanisms: It helps in pinpointing the cellular origins of diseases by identifying dysfunctional cellular processes in individual cells.
- Developmental Biology: Researchers use scRNA-seq to trace the lineage and developmental pathways of cells in an organism, providing insights into embryonic development and tissue regeneration.
Benefits for Biologists:
- Enhanced Resolution: scRNA-seq provides a high-resolution view of cellular complexity, offering detailed insights that are not possible with bulk RNA sequencing where signals from multiple cells are averaged.
- Targeted Research: This method allows biologists to focus on specific cells of interest, such as those involved in disease or those exhibiting unique behaviors within a population.
- Dynamic Analysis: scRNA-seq is ideal for studying dynamic processes, like how cells respond to a treatment or environmental change over time.
Practical Applications in Research:
For biologists, scRNA-seq opens up new avenues for research, from cancer biology to immunology, by providing a detailed map of gene expression patterns at the single-cell level. It enables the discovery of new cell types, the understanding of complex cellular ecosystems, and the development of targeted therapies.
Getting Started with scRNA-seq:
While scRNA-seq offers profound insights, mastering this technique can be challenging due to its technical complexity and the advanced analysis required. For researchers eager to integrate single-cell sequencing into their projects, starting with a clear research question and a well-planned experimental design is essential. Additionally, leveraging workshops and training sessions on single-cell technologies can provide the necessary skills and knowledge to navigate this advanced field effectively.
Single-cell RNA sequencing is transforming our understanding of biology at the most fundamental level—cell by cell. As this technology continues to evolve, it promises to unlock even more secrets of life, one cell at a time, paving the way for groundbreaking discoveries in numerous fields of biological science. Embrace this cutting-edge tool and propel your research into new dimensions.
Harnessing Nextflow for High-Throughput Genomic Analysis: A Must for Large-Scale Studies
Posted on April 18, 2024

In today’s fast-paced scientific world, genomic research is expanding at an unprecedented rate. Handling and analyzing vast datasets effectively is essential, especially when dealing with large-scale studies. This is where Nextflow comes into play—an efficient, flexible tool designed to meet the challenges of modern genomics. Let’s break down what Nextflow is, its capabilities, and why it's becoming a cornerstone in genomic research.
What is Nextflow?
Nextflow is a workflow management system that enables scientists and bioinformaticians to deploy complex data analysis workflows. It's built around a programming language that combines conventional scripting with the robustness needed for high-throughput computing tasks. Nextflow simplifies the building and executing of scalable and reproducible scientific workflows across multiple platforms like local machines, cloud services, and high-performance computing environments.
Capabilities of Nextflow:
- Pipeline Scripting: Allows users to write workflows using a simple, domain-specific scripting language.
- Container Integration: Supports Docker, Singularity, and other container technologies to encapsulate workflows, ensuring they can be run in the same environment across all computational systems.
- Parallel Execution: Automates the distribution of tasks across available computing resources, optimizing performance and reducing processing time.
Benefits for Beginners and Advanced Users Alike:
- Ease of Use: Despite its powerful features, Nextflow is accessible for beginners. The syntax is straightforward, and the community provides extensive documentation and examples to help new users get started.
- Reproducibility: By using containers, Nextflow ensures that all computational environments are consistent, making scientific results reproducible no matter where the workflow is executed.
- Scalability: Whether you’re analyzing data from a few samples or handling large cohorts involving thousands of genomes, Nextflow scales effortlessly with your needs.
- Community Support: A vibrant community of users contributes to a growing library of shared workflows, offering support and innovation.
Practical Application in Genomic Research:
For a biologist embarking on genomic analysis, Nextflow provides the tools to not only manage data more efficiently but also to enhance the quality of their research. It allows for the standardization of analysis protocols, making studies more straightforward to replicate and validate.
Taking the Next Step with ecSeq Workshops:
Understanding and using Nextflow effectively can significantly benefit your research, and ecSeq’s specialized workshops are designed to help you achieve just that. Our Nextflow workshops provide practical, hands-on training tailored to the needs of genomic researchers. Participants learn to construct, run, and manage their workflows, gaining the confidence to tackle complex genomic analyses.
For anyone involved in large-scale genomic studies, learning Nextflow through ecSeq can be a transformative experience, enabling you to handle your projects more professionally and with greater ease.
Explore the potential of Nextflow and enhance your research capabilities by participating in one of our comprehensive Nextflow workshops. Embrace the future of genomic analysis and take your research to new heights with the power of Nextflow. Join us to discover how this tool can revolutionize your approach to genomic data processing.
RNA-Seq Data Analysis Workshop, March 2024
Posted on March 18, 2024

The recent RNA-Seq Data Analysis Workshop in Berlin, spanning March 11 - 14, 2024, offered an immersive experience in RNA-Seq data analysis. With 13 participants from Denmark, Spain, Germany, Romania, Poland, the UAE, and the Czech Republic, the workshop highlighted its international draw. Attendees engaged in extensive hands-on sessions, leveraging state-of-the-art tools to dissect and interpret RNA-Seq data, a process that not only bolstered their analytical skills but also prepared them for collaborative and independent research endeavors in the dynamic field of genomics.
The course is designed to bridge the knowledge gap in NGS data analysis, providing researchers with the tools and confidence needed to embark on their bioinformatics journey. For insights into the participants' experiences and their feedback on the course, please visit here.
Online Course - A Practical Introduction to NGS Data Analysis, February 2024
Posted on March 05, 2024

The recent online course titled "A Practical Introduction to NGS Data Analysis," held from February 28 to March 1, was an engaging and in-depth exploration into the world of NGS data analysis. This session attracted 22 participants from a diverse array of countries, including Germany, Denmark, Australia, Poland, Japan, Iceland, the UK, the USA, Canada, the United Arab Emirates, and Lithuania, showcasing its global appeal.
Over the course of three days, participants delved into the fundamentals of NGS data analysis, gaining hands-on experience through our user-friendly, browser-based terminal. This practical approach allowed attendees to apply their new knowledge in real-time, analyzing their first data set by the end of the course. Such immediate application is invaluable, empowering researchers to begin working on their own data sets and enhancing their ability to engage in meaningful discussions with bioinformaticians.
The course is designed to bridge the knowledge gap in NGS data analysis, providing researchers with the tools and confidence needed to embark on their bioinformatics journey. For insights into the participants' experiences and their feedback on the course, please visit here.
Unlocking Genomic Insights with Personalized Software Solutions
Posted on February 01, 2024

In the rapidly advancing realm of genomics, the ability to extract meaningful insights from vast amounts of genetic data is crucial for unlocking new frontiers in medicine and research. ecSeq Bioinformatics, a leading provider of custom NGS analysis software development, empowers researchers and clinicians to achieve this goal by crafting tailored solutions that seamlessly integrate into their workflows and deliver exceptional results.
Understanding and Aligning with Client Needs
Our journey begins with a deep understanding of our clients' specific needs and challenges. We conduct in-depth discussions, analyzing their unique research objectives, data requirements, and technological constraints. This meticulous approach ensures that our software is not merely functional but rather a powerful tool that becomes an integral part of their research and diagnostic strategies.

Delving into the Depths of Genomic Complexity
We go beyond superficial analysis to delve into the intricate details of each project. This includes understanding the diverse nature of genomic data, its implications for interpretation, and the specific analytical challenges it presents. By thoroughly grasping the complexities of genomic studies, we ensure that our software is tailored to address the exact needs of each client.
Collaboration: The Foundation of Innovation
We foster a collaborative environment where our expertise meets the insights of our clients. This open dialogue ensures that the software development process is a shared journey, leading to innovative solutions that are both effective and forward-thinking.
Seamless Integration and Adaptability
Our solutions are designed to integrate seamlessly into existing laboratory ecosystems. We prioritize compatibility and workflow efficiency, allowing researchers to integrate our software into their existing processes without disruptions.

Quality and Precision: The Hallmarks of Excellence
At ecSeq Bioinformatics, quality and precision are non-negotiable. We subject our software to rigorous testing and validation procedures, ensuring that each iteration meets the highest standards of scientific accuracy and reliability. Our commitment to excellence sets us apart in the world of genomic software development.
Security and Confidentiality: Protecting Sensitive Information
In a world where data security is paramount, we take robust measures to safeguard sensitive genetic information. Our software is fortified with state-of-the-art security protocols, providing our clients with the peace of mind that their data is protected with utmost care.
Feedback and Iteration: Continuous Improvement
We actively solicit feedback from our clients, incorporating their insights into ongoing development cycles. This iterative approach ensures that our software remains at the forefront of genomic research, constantly evolving to meet the changing needs of our clients and the ever-advancing field of genomics.

Empowering Research: Our Ultimate Goal
Our ultimate goal is to empower researchers and clinicians to make groundbreaking discoveries in the field of genomics. By providing them with powerful and intuitive software tools, we enable them to extract deeper insights from genetic data, pushing the boundaries of human knowledge and advancing the frontiers of medical research.
Unleash the Power of Personalized Software Development
If you are ready to revolutionize your genomic research and diagnostics, ecSeq Bioinformatics is here to help. Explore our comprehensive insights at our knowledge bank and discover how our bespoke software solutions can empower your work and lead to transformative breakthroughs.

Introduction to NGS Data Analysis Workshop, November 2023 in Munich
Posted on December 05, 2023

Our recent Next-Generation Sequencing Data Analysis workshop in Munich, held from November 29 to December 1, was a highly engaging and informative event. We were delighted to host 13 participants from an impressive array of countries, including Norway, the Netherlands, the United States, Slovenia, Belgium, Italy, Qatar, Oman, Poland, and Germany.
With the expert guidance of our trainers, the participants delved into the world of NGS data analysis. The workshop provided a hands-on introduction to the field, allowing attendees to gain a deeper understanding of the underlying concepts and tools used in NGS. They learned practical skills such as working with the command line and analyzing sequencing data, which are invaluable for their future research endeavors.
The diverse backgrounds of the participants added a unique dimension to the workshop, fostering excellent opportunities for networking and discussions. Overall, the event was not just a learning experience but a platform for professional growth and collaboration.
This workshop in Munich has further established our commitment to providing top-quality education in bioinformatics. For those looking to expand their knowledge and connect with other professionals in the field, our workshops offer an unparalleled opportunity. Stay tuned for our upcoming courses and join us in this exciting journey of discovery and learning!
Bioinformatics Pipeline Development with Nextflow (November 2023, online)
Posted on November 20, 2023

Our recent Bioinformatics Pipeline Development with Nextflow online course, held last week, marked a significant milestone in our journey of providing top-tier bioinformatics education. We were delighted to host 18 dedicated participants from Serbia, the USA, Germany, Spain, and Austria, bringing together a rich tapestry of perspectives and experiences.
Under the guidance of our skilled trainers, each attendee was immersed in the world of Nextflow, gaining hands-on experience and a profound understanding of bioinformatics pipeline development. This course was more than just an educational session; it was a transformative experience that equipped participants with the tools and confidence to design and implement their own bioinformatics workflows.
This workshop has raised the bar in enabling researchers and bioinformaticians to master the intricacies of Nextflow and pipeline development. If you're eager to expand your bioinformatics skill set and join a community of forward-thinking professionals, our workshops are your gateway. Keep an eye out for our upcoming courses and seize the opportunity to be part of our next groundbreaking event!

Single-Cell RNA-Seq Data Analysis Workshop, Nov 2023 in Berlin
Posted on November 13, 2023

Our inaugural single-cell RNA-Seq workshop in Berlin, spanning from November 8-10, was a phenomenal hit! We were thrilled to welcome 24 eager learners from a diverse array of 7 countries - Germany, Denmark, Poland, Saudi Arabia, the UK, Thailand, and France. This global mix of participants added a unique vibrancy to our course.
Guided by our trio of expert trainers, each participant received personalized attention, ensuring a deep and practical understanding of Linux and single-cell RNA-Seq data analysis. The workshop wasn't just about learning new skills; it was an immersive experience where participants gained the confidence and competence to independently conduct their analyses and engage in insightful discussions with bioinformaticians.
This workshop has set a new benchmark in empowering researchers to confidently navigate the complexities of bioinformatics. If you're looking to enhance your skills and join a growing community of proficient bioinformaticians, our workshops are the perfect opportunity. Stay tuned for our upcoming courses and join us in this exciting journey of discovery and learning!
Learn more about our future workshops and be part of our next success story!
The Unmatched Value of ecSeq Bioinformatics Workshops
Posted on November 09, 2023
Unlocking the World of Bioinformatics: A Gateway for All
At ecSeq Bioinformatics, we've crafted our workshops to be a melting pot of knowledge and experience, where professionals from diverse backgrounds - molecular biologists, medical doctors, pharmacists, pathologists, and many others - converge to explore the dynamic world of bioinformatics. What makes these workshops truly unique is their accessibility; no prior knowledge of bioinformatics is required, making them an ideal starting point for anyone keen to delve into this field.

Learning by Doing: An Interactive and Hands-On Approach
Our workshops are designed around the philosophy of 'learning by doing'. Participants are not just passive listeners; they actively engage in typing and executing commands themselves, rather than merely copying and pasting. This hands-on approach ensures a deeper understanding and retention of the skills being taught. To facilitate this, we have assistants on-site, ready to offer help and guidance, ensuring a smooth and productive learning experience for everyone.

Beyond Learning: Networking and Discussions
One of the most valued aspects of our workshops is the opportunity they provide for networking. Breaks and meal times buzz with lively discussions, allowing participants to share insights, experiences, and forge professional connections. These interactions often lead to great discussions, sparking new ideas and collaborations.

A Certificate of Achievement and Its Impact
Upon completion of the course, participants receive a certificate, a testament to their newly acquired skills in bioinformatics. These certificates carry significant weight, as evidenced by the numerous inquiries we receive from companies. Many of our participants have successfully used these certificates to enhance their CVs and bolster their job applications, showcasing their commitment to continuous learning and professional development.

An Escape for Focused Learning
Our workshops offer more than just learning; they provide an escape from the daily grind. Hosted in various great cities, they allow participants to immerse themselves entirely in the course, free from everyday distractions. This focused environment is conducive to learning and understanding complex concepts more effectively.

The Added Bonus: Travel and Exploration
An added perk of attending our workshops is the opportunity to travel. Participants not only gain valuable knowledge but also get the chance to explore new cities, indulge in local cultures, and enjoy some sightseeing. This blend of education and exploration makes our workshops a memorable and enriching experience.

In conclusion, the ecSeq Bioinformatics workshops are more than just educational courses; they are a comprehensive experience that combines learning, networking, and exploration. Whether you're a beginner or a professional looking to expand your skill set, our workshops offer a unique opportunity to dive into the world of bioinformatics, make valuable connections, and explore new horizons. Join us on this exciting journey of discovery and growth!
Online Course - A Practical Introduction to NGS Data Analysis, October 2023
Posted on October 26, 2023
The recent online course titled "A Practical Introduction to NGS Data Analysis" was a comprehensive and rigorous program. Spanning three days, it catered to a substantial cohort of 24 participants, providing them with an initial understanding of NGS data analysis. Utilizing our browser-based terminal, participants were able to immediately implement the knowledge acquired and analyze their inaugural data set. This equips researchers with the capability to commence analysis on their personal data and facilitates informed discussions with bioinformaticians, bridging the knowledge gap. If you're interested in their feedback on the course, you can find it here.
7th Berlin Summer School in NGS Data Analysis, September 2023
Posted on September 12, 2023

What a wonderful week! The group that was at our summer school last week was just awesome. Nice and curious people, great questions, lots of interaction and most of all a lot of fun. At dinner and during the city tour there was a lot of talking, laughing and discussing. Just the right thing to be able to digest the acquired knowledge well. And thanks to the great weather, we were able to sit outside for a long time in the evening, go for a walk or do some sports to clear our heads again. An unforgettable week!
This time, 29 participants from 11 different countries (Austria, China, Denmark, Estonia, Germany, Italy, New Zealand, Norway, Romania, Slovenia, USA) attended our summer school in Berlin.
Are you also interested in participating in one of our courses? Then take a look at the courses we offer! We would be happy to welcome you with us.
RNA-Seq Data Analysis Workshop for SaxoCell
Posted on September 11, 2023

At the end of August, we were privileged to train a group of highly talented graduate students, postdocs and scientists from SaxoCell in the analysis of RNA-Seq data.
SaxoCell® is one of the winners of the innovation competition "Clusters4Future" of the Federal Ministry of Education and Research (BMBF).
The creation of the future cluster is based on an association of leading research institutes and medical facilities from Saxony, including Leipzig and Dresden.
Together with numerous other partners from academia and industry, they have set themselves the goal of opening up new areas of application and production methods for cell and gene therapeutics by means of twelve innovative research and development projects, among other things.
In addition, SaxoCell® will strengthen regional networking within the research area and technology transfer for the benefit of sustainable, local value creation.
ecSeq is part of the SaxoCell® project SaxoCellOmics. This project is addressing optimal and early monitoring of the development and production of gene and cell therapeutics. SaxoCellOmics pursues four strategic goals for the cluster: (1) the provision of efficient and harmonized processes for monitoring the delivery and tolerability of novel therapies, (2) the identification of mechanisms of action, new targets and resistances, (3) the development of improved quality criteria for the manufacturing process, and (4) the development of predictive biomarkers.
Find more information about the research project and ecSeq's involvement on the SaxoCellOmics website.
Join the NGS Revolution with On-Site Workshops for Research Groups
Posted on July 04, 2023

In the dynamic world of genomics and bioinformatics, staying at the forefront of scientific progress is essential for groundbreaking research. As researchers, we understand the challenges and opportunities that come with next-generation sequencing (NGS) data analysis. That's where ecSeq's on-site NGS data analysis workshops come in, providing customized training to enhance the capabilities of your research group. In this blog post, we explore why ordering such a course can be a game changer, opening new horizons for your scientific journey. 🌟
Customized Curriculum for Research Excellence:
Imagine having the ability to tailor the curriculum to perfectly match the goals and interests of your research group. With ecSeq's on-site workshops, this becomes a reality. Whether you are researching genetic testing, studying biodiversity, or diving into epigenetics, trainers will work with you to create a customized curriculum that addresses your specific research needs. This personalized approach ensures that the knowledge gained during the workshop directly contributes to advancing your research projects and promoting your scientific success. ✨
Expert Trainers Fueling Discovery:
At ecSeq, instructors are more than just trainers; they are highly experienced researchers themselves. Their expertise spans a broad range of NGS data analysis areas, ensuring that you are guided by professionals with first-hand knowledge of the field. Their practical insights, honed through academic and industry experience, equip your research group with the skills and tools needed to overcome challenges and achieve breakthroughs. With their help, you can embark on your scientific journey with confidence and discover new areas of knowledge. 🔬
Embracing Open-Source Tools for Collaboration:
In today's scientific landscape, collaboration and accessibility are paramount. The ecSeq workshops emphasize the use of open source tools that are freely available to the scientific community. By using these tools, your research group can seamlessly collaborate with other institutes, share methods, and contribute to the broader scientific community. In addition, instructors will introduce various software options so that you can choose the most appropriate tools for your research goals. This collaborative and open approach promotes knowledge sharing and enhances the impact of your research. 🤝
Comprehensive Learning Materials for Ongoing Success:
ecSeq's commitment to your success extends far beyond the workshop itself. The carefully prepared learning materials and sample data sets provided during the course serve as invaluable resources for your ongoing research. These materials enable your research group to effectively apply the concepts and techniques learned to ensure that the knowledge gained during the workshop lingers long after its completion. With these curated resources, you can confidently tackle new challenges, refine your analyses, and advance your research. 📚
Convenient On-Site Training for Efficiency and Growth:
When you choose on-site workshops, you reap the benefits of convenience, efficiency, and growth for your research group. There is no need for long travel or interruptions to your research schedule. Instructors come to your institute, creating a focused and intensive learning environment in the familiar surroundings of your research facility. This saves you valuable time, avoids travel expenses, and allows you to use your resources more effectively, ultimately maximizing your research results and scientific progress. ⏰💰
Contac Us Now:
Investing in ecSeq's customized on-site NGS data analysis workshops is an investment in your research group's success. By customizing the curriculum, benefiting from experienced trainers, using open-source tools, accessing comprehensive learning materials, and enjoying the convenience of on-site training, you empower your research group to push boundaries, make significant discoveries, and contribute to the advancement of scientific knowledge. Take advantage of this opportunity, share this information with your colleagues and mentors, and embark on a transformative journey of scientific exploration and achievement. 🚀🔬
Learn more about our offer of on-site courses.
Inspecting Consumer Whole-Genome Sequencing Data
Posted on June 15, 2023

As a provider of software, services and training in the area of next generation sequencing (NGS) bioinformatics, we naturally see a lot of sequencing data. The “end users” of our analysis results are typically life science researchers, or biotech- or medical professionals. Recently, a new breed of companies started offering comprehensive DNA sequencing for consumers. For example, they might offer Whole-Genome-Sequencing (WGS), at a reasonable level of coverage, together with a set of potentially interesting analyses, all at an incredibly low price of $300. Therefore, we could not resist the temptation and ordered such a test with the overall goal of checking the amount and quality of data obtained.
What you receive
We decided to order a DNA test from Nebula Genomics, which is one of the largest providers that also offers downloading of the genomic data. After sending back the cheek swab, we had to wait for several weeks until the results were in. You are now able to inspect a range of reports about your genetic traits (simple and complex), ancestry, oral microbiome and genetic variation. A deep-dive discussion of these reports is a little out of scope for what we want to do here, and can be found elsewhere already. Of more immediate interest to us is the raw data itself, which underpins all downstream analysis. We directly went on to the data download section, and were positively surprised about the specific files offered for download. Before ordering, we checked the FAQ, which stated that we will be able to download all data in “BAM format” and “VCF format”, both important for describing sequence alignment characteristics and genetic variant information, respectively. Since BAM files contain alignments to the human reference genome, it was not clear from the FAQ however if the full dataset - including also sequences not aligning to the reference - would be provided.
We now found that we were able to download:
- two FASTQ files, containing the all raw sequence reads
- a CRAM file, containing the alignments to the reference genome in a highly compressed file format (but with the same information as in a BAM file)
- a VCF file, containing the detected DNA variants
- Corresponding index files for the CRAM and VCF files which are helpful to increase the performance when working with alignments and variants, respectively
In other words, we are given the complete data in standardized file formats.
It’s worth noting that Nebula Genomics does not provide any QC reports alongside this data. In order to assess the provided data we must analyze it ourselves. So, the following visualizations were created with our own Seamless NGS software and open source tools. Nebula Genomics also does not provide a description of their data analysis workflow, so we will have to reverse engineer some of this information based on the files provided.
Quantity and quality of raw sequencing data
We first looked at the raw sequencing data from the provided FASTQ files:
- Sequences : 2x 382,683,421
- Sequence length: 150 bp
- Total data: 2x 57,4 Gbp
- File sizes: 2x 52 GiB
The distribution of base qualities is usually one of the most important quality metrics to look at:
We can observe that the medium base quality for all positions in the read sequence averages to a median of 36 (Phred score). When we average the quality for all bases in the read, we see that most sequences have a mean quality score of 37, while some even go up to 39. This amounts to a very good quality by today's standards. According to their FAQ, they employ the MGI DNBSEQ-T7 sequencing machine. Most data from the market leading Illumina sequencing machines will report base qualities that top at 36. Does that mean that the quality here is better than from those common Illumina machines? Not really, since the quality scores reported are essentially estimated by an internal software that has been calibrated by the vendor and any comparison between different sequencing machines should not be based on these numbers. More in-depth studies (e.g. Hak-Min Kim et al) suggest that the raw sequence quality is roughly comparable between those most common platforms. What this means however is that the sequencing laboratory did a good job in operating the sequencing machine.
Alignments
Nebula genomics provides a CRAM file containing the sequences aligned to a reference genome. Let us first find out what tool they used for this step, and what reference. Luckily, the CRAM file itself contains this information: they used the alignment tool minimap2. This is a bit unusual in that bwa-mem is the more widely-used tool, despite being its predecessor, and also the standard tool recommended for the variant analysis pipeline employed later on. However, for most data minimap2 has been shown to provide similar accuracy for variant detection while providing a significant runtime speedup compared to bwa-mem. The output format is quite similar. With the expectation that MGI made some internal tests for how well it works for the MGI DNBSEQ-T7 sequences, this tool is a sensible choice given that they have to handle high data volumes.
The CRAM file also reveals some information regarding what reference genome they have been using. This can be reverse-engineered based on the names and checksums of the chromosomes, for example. This reference version information is of great importance, because if we aim to do any additional analysis using the result files provided by Nebula Genomics Inc., we need to ensure that the same reference genome is used. The reference genome turns out to be a modified version of the Genome Reference Consortium Human Build 38 (GRCh38) using UCSC nomenclature. All information points to the fact that they used Verily's GRCh38 genome, but when comparing it to the downloadable version we found that there are some mismatches in sequence checksums. We asked Nebula for the precise reference genome, but unfortunately they did not provide any answer.
We could however infer that the reference is based on the so-called “analysis set” of hg38. The hg38 contains several features that complicate data analysis (find some details here), and these special versions of the GRCh38 reference genome facilitate a correct use of common read mapping tools with hg38. Notably, the names and coordinates of the 23 chromosomes are the same as the UCSC hg38 assembly, meaning that you should be able to use the VCF file with any hg38-based analysis.
Let’s take a brief look at the alignment statistics:
As expected, most of the reads - 99,4 % to be precise - could be aligned successfully to the human reference genome. This is a very good number, and it means that there was no major contamination of the sample which can happen, for example if you have a viral infection while taking the mouth swabs. The amount of DNA collected on the swab can also be a bottleneck for sequencing and it's good to see that this wasn't an issue here.
Using the information about known gene locations, we can also look where the reads actually map to:
Human gene sequences consist of introns and exons; the latter are translated into proteins that carry out most functions in the cell. For humans, these “exonic regions” amount to 1-2% of the genome. As you can see, 2% of the reads align to exonic regions, which is the amount we expect. The majority of reads however, align to either intronic or intergenic regions.
Where are the bacterial reads?
One of the reports provided is about the oral microbiome, so the types and abundances of bacteria that were present in your mouth at the time of testing. But how can they know that when they align it only to the human genome? Well, indeed it is not possible to estimate that from the alignments that you get. But. If you look again on our mapping statistics above, you see that 0.6% of the reads do not align with the human genome. And since we are really looking at a lot of data, these 0.6% amount to almost 5 million sequences that we can use for further analysis. Fortunately, Nebula Genomics chose to include the unmapped reads in the CRAM file, which makes it easy to extract them and conduct further microbial analysis of your own! We used the well-known taxonomic classification tool Kraken2 and indeed obtained results very similar to their oral microbiome report.
Coverage statistics
When it comes to the assessment of sequencing data quality, a key question is: how good is the obtained coverage? Coverage means (link article) how many read sequences “cover” each position in our genome, providing us with a sufficiently large sample size to confidently detect changes. Nebula Genomics promised a coverage of 30x. Let us check if they can fulfill this promise:
The average coverage for the complete genome is 36.1, exceeding the expected value by about 20%. Very nice. We also see that there is some variation between the chromosomes. The biggest outliers are chrX and chrY, which is expected as I have only one of each compared to two of the others… It is a bit unfortunate that the chrY coverage is only 11.9 because this information is valuable for ancestry analysis. I left out the mitochondrial genome (chrM), because with a coverage of 9,620 this is a massive outlier! This is also expected, as each cell contains hundreds to thousands of copies of mtDNA.
Let us consider that above numbers represent averages over all possible positions in the genome. In practice, the coverage is not uniform but rather looks like this:

We see that there are coverage hills and valleys, and sometimes the coverage also goes down to 0. The reason is that certain regions of the genome differ in their amenability for sequencing according to their specific sequence content (see here). Unfortunately there is no real way to address this issue, other than by increasing the global level of sequencing depth to ensure that most regions still reach a minimum level of coverage. Ideally, we would like to get a coverage of 20x or more for all positions, because this allows us to confidently detect the germline variants for those positions. We can detect variants also at lower coverages, but with lower confidence. It is likely for this reason that Nebula Genomics offers a global coverage of 30x as a tradeoff.
When it comes to health-related information, often the most directly-relevant regions in the genome are those that eventually get made into proteins and can have a function in the cell. Naturally, it is interesting to look at how the coverage is in this high-priority region. So we did the analysis again, this time limiting ourselves to exonic regions, amounting to about 35 megabases (using a slightly modified set of RefSeq genes, similar to those used for Exome enrichments). Here is how the coverage looks like:
The mean coverage in this region is 32X, and 88% of positions have a coverage above 20x. Unfortunately, about 1 megabase (2.7% of those positions) have such a low coverage that it was not possible to detect variants at all. However, to put this in perspective, also in much more expensive clinical exome NGS kits between 1-2% of positions cannot be called. So this is something that must be expected and can only be resolved with more expensive techniques.
DNA Variants
The last file that we are offered to download contains the detected DNA variants in VCF format. For our sample, the total number was a sweeping 4.84 million variants. About 27k (or 0.6%) of these variants were located in exonic regions. This fraction is significantly less than the fraction of the exonic regions compared to the whole genome. This is entirely expected, since protein-coding exons have a more direct influence on biological processes, and are naturally more conserved due to natural selection. Note that many of the GWAS-related variants are actually outside of exonic regions because of the way these studies are performed.
To detect the variants, Nebula Genomics relied on the GATK HaplotypeCaller as part of the MGIs. HaplotypeCaller is the most widely-used tool for detecting DNA variants from NGS data, and it is completely open source. This is a good choice because it makes it easy to interpret the results and also increases the interoperability with other tools. The whole workflow includes steps for marking duplicates, recalibrating base quality scores and sorting the alignment files, all of which are key steps of a best practices workflow.
Duplication refers to the occurrence of the exact same sequence multiple times. Despite the huge amount of short-read data we are sequencing, the likelihood of such duplicates occurring naturally is in fact very low. High levels of duplication are therefore indicative of issues during sample preparation. Due to the biases introduced by duplicates, high levels can result in reduced confidence in the accuracy of variant calls. Consequently, they are typically excluded from variant detection. We independently assessed the duplication levels, and discovered that there were 24 million (3.2%) duplicate sequences in the data. This aligns with expectations based on other samples of good quality.
Thankfully the VCF already includes a DBsnp identifier. DBsnp is the largest database of human SNPs and having a pointer to the respective database entries simplifies working with the variants a lot. In our case, an identifier was available for about 97% of the detected variants.
Summary
With this we conclude our analysis of the WGS data you can get from consumer genetic testing company Nebula Genomics Inc. The sequencing data comes in sufficient quantities and also in very good quality. Together with the read length of 150 bp, which is good by current short-read sequencing standards, this means that most sequences can be aligned to the human genome, enabling the detection of a high number of DNA variants. Furthermore, Nebula Genomics deserves kudos for not only making available the most relevant files (including the complete raw sequences) but also for using standard tools (and consequently known standard contents) to generate these files. It would be possible to provide some additional QC metrics and analysis results (such as structural variations), and this is partly done by some competitors. However it is totally understandable for Nebula Genomics to not provide them, since in fact you need to be an expert to interpret those results properly - and an expert could also create them for yourself given sufficient computing resources. In summary, I am positively surprised about the great data you can get today for such a small cost. How much actionable advice and learnings you - as an consumer - are able to get from this data by today's knowledge, however, is a different story.
In-Person Workshop - Bioinformatics Pipeline Development with Nextflow, May 2023
Posted on May 19, 2023

Our Bioinformatics Pipeline Development with Nextflow workshop in Leipzig, Germany, held from May 15-17, was a highly productive and engaging event. We were thrilled to welcome 6 participants from 3 different countries: Germany, Italy, and the UK.
During the workshop, our expert trainers guided the participants through the process of developing bioinformatics pipelines using Nextflow. Through hands-on exercises and practical examples, the participants gained valuable skills and insights into building efficient and reproducible workflows. The workshop fostered collaborative discussions among the participants, allowing them to exchange ideas and experiences in pipeline development.
By the end of the workshop, the participants were equipped with the knowledge and tools to design and customize their own bioinformatics pipelines using Nextflow. They left the event with a solid foundation to tackle complex data analysis challenges and contribute to the advancement of bioinformatics research in their respective fields.
If you're interested in their feedback on the course, you can find it here.
Introduction to NGS Data Analysis Workshop, May 2023 in Munich
Posted on May 08, 2023

Our Next-Generation Sequencing Data Analysis workshop in Munich from May 3-5 was a practical and informative event. We had 15 participants from two different countries, namely Germany and Belgium.
With the guidance of our trainers, the participants received a hands-on introduction to NGS data analysis, allowing them to gain a deeper understanding of the underlying concepts and tools used in the field. They learned practical skills such as working with the command line and analyzing sequencing data, which will be invaluable for their future research endeavors. Overall, the workshop provided an excellent opportunity for participants to expand their knowledge and engage in discussions with other professionals in the field.
RNA-Seq Data Analysis Workshop, March 2023 in Berlin
Posted on April 03, 2023

The RNA-Seq workshop held in Berlin from March 27-30 was a resounding success. We had 24 participants from 7 different countries (France, Spain, Croatia, Slovenia, Germany, UK, and the Netherlands) who attended the course.
With the assistance of three trainers, we were able to provide excellent support to all 24 participants. They received a comprehensive overview of Linux and RNA-Seq data analysis, which equipped them with the knowledge necessary to carry out their own analyses and engage in meaningful discussions with bioinformaticians. This will allow them to tackle bioinformatics problems and questions with greater confidence and proficiency.
Online Course - A Practical Introduction to NGS Data Analysis, February 2023
Posted on February 06, 2023

Our online course “A Practical Introduction to NGS Data Analysis” last week was an intensive event. The relatively small group of 10 participants was able to get a first insight into the world of NGS data analysis during the three days. By using our browser-based terminal, they directly applied the knowledge gained and analyzed their first data set. This allows the researchers to directly start analyzing their own data and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level. If you're interested in their feedback on the course, you can find it here.
Introduction to NGS Data Analysis Workshop, Dec. 2022 in Munich
Posted on December 15, 2022

Our Introduction to NGS Data Analysis Workshop in Munich this week was a great event. The relatively small group of 14 participants was incredibly curious, interested and hardworking. Great questions and lots of discussions always lighten up the courses! The researchers got an introduction on how to work with linux and how to start analyzing RNA-Seq and DNA-Seq data. The gained knowledge will allow them to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
RNA-Seq Data Analysis Workshop, Oct. 2022 in Leipzig
Posted on October 28, 2022

Our RNA-Seq Data Analysis Workshop in Leipzig this week was a great event. We were able to help the 20 participants perfectly because we were there with three trainers. The researchers got an introduction on how to work with linux and how to analyze RNA-Seq data correctly. The gained knowledge will allow them to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
In-Person Workshop - Bioinformatics Pipeline Development with Nextflow, October 2022
Posted on October 10, 2022

Our in-person course “Bioinformatics Pipeline Development with Nextflow” last week was a great event. The 11 participants from 4 different countries (Belgium, Germany, Saudi Arabia and Norway) learned how workflow management systems can accelerate their research. By using a linux terminal, they were able to directly apply the knowledge gained and together implement and start their first pipeline with Nextflow. If you're interested in their feedback on the course, you can find it here.
Online Course - A Practical Introduction to NGS Data Analysis, September 2022
Posted on October 04, 2022
This week we held our last online intro course this year. Once again it was a great event and the 21 participants from 11 countries (Saudi Arabia, Italy, Switzerland, Germany, Hungary, Finland, USA, Netherlands, Austria, Denmark, Egypt) made it through the three packed days. Of course, the partly very big time difference made some participants uncomfortable, but it saves long flights a jetlag. We want to thank all participants for their trust. If you're interested in their feedback on the course, you can find it here.
Doctoral Thesis with Honors at ecSeq
Posted on September 07, 2022

Adam Nunn successfully defended his doctoral thesis on September 5, 2022
On Monday, our PhD student Adam Nunn successfully defended his thesis at the Faculty of Mathematics & Computer Science of the Leipzig University.
The referees all agreed in their decisions: "Summa cum laude" (Passed with highest honors)
Adam has achieved incredible things in the last four years. He has managed the complete server infrastructure of the Marie Skłodowska-Curie Innovative Training Network EpiDiverse, was the main contact person for all 15 PhD students of the project when it came to bioinformatics questions and assembled a new genome in cooperation with University of Minnesota. He also designed, created and maintained several methylation pipelines, which were used by all PhD students for their research.
And here's the thing: He did all of this in parallel to his PhD work.
His PhD thesis was on detecting DNA variants in Bisulfite-Seq data. For this, Adam chose an incredibly simple and clever approach that allowed him to outperform all the tools already on the market. The best inventions don't always have to be the most complicated. Simplicity is where true innovation lies.
During his time as a graduate student, Adam published six papers and was first author on four of these:
-
Nunn A, Otto C, Fasold M, Stadler PF, Langenberger D: 'Manipulating base quality scores enables variant calling from bisulfite sequencing alignments using conventional bayesian approaches', BMC Genomics 23, Article number: 477 (2022)
-
Nunn A, Rodríguez-Arévalo I, Tandukar Z, Frels K, Contreras-Garrido A, Carbonell-Bejerano P, Zhang P, Ramos-Cruz D, Jandrasits K, Lanz C, Brusa A, Mirouze M, Dorn K, Galbraith D, Jarvis BA, Sedbrook JC, Wyse DL, Otto C, Langenberger D, Stadler PF, Weigel D, Marks MD, Anderson JA, Becker C, Chopra R: 'Chromosome-level Thlaspi arvense genome provides new tools for translational research and for a newly domesticated cash cover crop of the cooler climates', Plant Biotechnol J., 20(5):944-963 (2022)
-
Gawehns F, Postuma M, van Antro M, Nunn A, Sepers B, Fatma S, van Gurp TP, Wagemaker NCAM, Mateman AC, Milanovic-Ivanovic S, Groβe I, van Oers K, Vergeer P, Verhoeven KJF: 'epiGBS2: Improvements and evaluation of highly multiplexed, epiGBS-based reduced representation bisulfite sequencing', Mol Ecol Resour, 22(5):2087-2104 (2022)
-
Nunn A, Can SN, Otto C, Fasold M, Díez Rodríguez B, Fernández-Pozo N, Rensing SA, Stadler PF, Langenberger D: 'EpiDiverse Toolkit: a pipeline suite for the analysis of bisulfite sequencing data in ecological plant epigenetics', NAR Genomics and Bioinformatics, 3(4), lqab106 (2021)
-
Can SN, Nunn A, Galanti D, Langenberger D, Becker C, Volmer K, Heer K, Opgenoorth L, Fernandez-Pozo N, Rensing SA: 'The EpiDiverse Plant Epigenome-Wide Association Studies (EWAS) Pipeline.', Epigenomes, 5(2):12 https://doi.org/10.3390/epigenomes5020012 (2021)
-
Nunn A, Otto C, Stadler PF, Langenberger D: 'Comprehensive benchmarking of software for mapping whole genome bisulfite data: from read alignment to DNA methylation analysis', Briefings in Bioinformatics, Volume 22, Issue 5 (2021)

Adam Nunn started with a BSc in Biological Sciences from University of Portsmouth and a double MSc in Bioinformatics and Biology from Lund University, Sweden. Between the bachelor's and the master's degree, he worked for four years as a lead research scientist for a UK-based company where he focused on developing biological alternatives to chemical pesticides. In 2017 he joined ecSeq Bioinformatics GmbH for his PhD studies. His work is fully funded by the Marie Skłodowska-Curie Innovative Training Network EpiDiverse and in cooperation with Prof. Peter F. Stadler, holder of the bioinformatics chair at Leipzig University. On September 5, 2022, he successfully defended his doctoral thesis at the University of Leipzig. His work was evaluated with a "Summa cum laude" and he will be entitled to use the title "Dr. rer. nat" in the future.






NGS data analysis workshops in the second half of 2022
Posted on August 01, 2022
NGS data analysis workshops in the second half of 2022
Finally it's possible to meet people and visit great cities again! We are offering face-to-face courses again in the second half of 2022, as we have learned to deal with the Covid-19 pandemic. Despite large groups, we had no cases of infection in our events. With the help of testing, precautions, and of course responsible participants, we were able to offer safe events.
Networking, eating together and going out for a drink in the evening. All of this is only possible if you go to classroom courses. Find a trade-off between safety and life. Attend one of our in-person courses in the second half of 2022!
Of course, we will continue to offer our intro course online. Here we can ensure that there are no disadvantages (apart from networking and perhaps quality of the lunch) to the face-to-face course.

Advance your research. Understand NGS and analyze sequenced data yourself.
In a nutshell:
- Learn the essential computing skills for NGS bioinformatics
- Understand NGS technology, algorithms and data formats
- Use bioinformatics tools for handling sequencing data
- Perform first downstream analyses for studying genetic variation

Streamline your research through the development of reproducible analysis pipelines.
In a nutshell:
- Learn the fundamental best-practices of bioinformatic pipeline development
- Understand how workflow management systems can accelerate your research
- Use state-of-the-art, open source software to make complex analyses routine
- Perform your own custom analysis pipelines using Nextflow!

Advance your research. Understand RNA-Seq analyses challenges and solve them yourself.
In a nutshell:
- Learn the essential computing skills for NGS bioinformatics
- Understand NGS analysis algorithms (e.g. read alignment) and data formats
- Use bioinformatics tools for handling RNA-Seq data
- Create diagnostic graphics and statistics
- Compare different approaches for differential expression analysis

Advance your research. Understand NGS and analyze sequenced data yourself.
In a nutshell:
- Learn the essential computing skills for NGS bioinformatics
- Understand NGS technology, algorithms and data formats
- Use bioinformatics tools for handling sequencing data
- Perform first downstream analyses for studying genetic variation
Clever Algorithm - Use standard DNA variant caller on Bisulfite-Seq data
Posted on June 27, 2022

Until now, specialised tools are required in order to access DNA variants from bisulfite sequencing data. Experts in the field are often frustrated by the output and performance of the few available variant callers for BS-seq data, to the extent that they tend to re-sequence an additional whole genome DNA-Seq run just to access prominent/trustable tools like GATK (McKenna et al., 2010) or Freebayes (Garrison & Marth, 2012). This results in an expensive outlay in terms of additional cost and labor.
Take a look at the simplicity of the method! It is both straightforward to understand for those outside the field and showcases how a small change in perspective can lead to novel and unexpected solutions.
Reference:
Nunn A, Otto C, Fasold M, Stadler PF, Langenberger D: 'Manipulating base quality scores enables variant calling from bisulfite sequencing alignments using conventional bayesian approaches', BMC Genomics 23, Article number: 477 (2022)

ecSeq Bioinformatics GmbH among the top 10 emerging NGS companies in Europe 2022
Posted on June 25, 2022

Great News - ecSeq Bioinformatics GmbH is among the top 10 emerging NGS companies in Europe 2022 - Life Sciences Review (Europe Special)
What a nice gift for our 10th anniversary! It's always great to see when years of work pay off! But without our great employees, this big step wouldn't have been possible.
Thanks also go to our many customers with whom we have had the pleasure of working over all these years.

1st SaxoCell® Consortium Meeting 2022
Posted on June 24, 2022
Living Drugs - Precision therapy cluster for Saxony
This month was the first face-to-face meeting of the SaxoCell® Consortium and finally, after all the months of planning via Zoom, we were able to meet in person. This great event highlighted once again how important it is to be face-to-face. New contacts were made, new ideas were born and new collaborations were formed. Networking at its best. And of course, the fun must not be neglected. A great time, a great event!

SaxoCell® is one of the winners of the innovation competition "Clusters4Future" of the Federal Ministry of Education and Research (BMBF).
The creation of the future cluster is based on an association of leading research institutes and medical facilities from Saxony, including Leipzig and Dresden.
Together with numerous other partners from academia and industry, they have set themselves the goal of opening up new areas of application and production methods for cell and gene therapeutics by means of twelve innovative research and development projects, among other things.
In addition, SaxoCell® will strengthen regional networking within the research area and technology transfer for the benefit of sustainable, local value creation.
ecSeq is part of the SaxoCell® project SaxoCellOmics. This project is addressing optimal and early monitoring of the development and production of gene and cell therapeutics. SaxoCellOmics pursues four strategic goals for the cluster: (1) the provision of efficient and harmonized processes for monitoring the delivery and tolerability of novel therapies, (2) the identification of mechanisms of action, new targets and resistances, (3) the development of improved quality criteria for the manufacturing process, and (4) the development of predictive biomarkers.
Find more information about the research project and ecSeq's involvement on the SaxoCellOmics website.



6th Berlin Summer School in NGS Data Analysis, June 2022
Posted on June 17, 2022

Finally a face-to-face course with many participants again! Our 6th Berlin Summer School in NGS Data Analysis in Berlin this week was just great. The 30 participants from 14 different countries (Croatia, Uruguay, Slovenia, Germany, USA, Italy, Slovenia, Serbia, Switzerland, Czech Republic, Norway, Netherlands, Poland, Ireland) had a great time and enjoyed the networking possibilty. The researchers got an introduction on how to work with linux and how to analyze DNA-Seq and RNA-Seq data correctly. What a great event.


Online Course - A Practical Introduction to NGS Data Analysis, May 2022
Posted on May 19, 2022
This week we held our second online intro course this year. Once again it was a great event and the 14 participants from 9 countries (Austria, Belgium, Germany, USA, Denmark, UK, Lithuania, Hong Kong, Netherlands) made it through the three packed days. Of course, the partly very big time difference made some participants uncomfortable, but it saves long flights a jetlag. We want to thank all participants for their trust. If you're interested in their feedback on the course, you can find it here.
Online Course - Bioinformatics Pipeline Development with Nextflow, May 2022
Posted on May 16, 2022
Our online course “Bioinformatics Pipeline Development with Nextflow” this week was a great event. The 12 participants from 19 different countries (Austria, Belgium, Germany, USA, Denmark, UK, Austria, Lithuania, Hong Kong, Netherlands) learned how workflow management systems can accelerate their research. By using our browser-based terminal, they were able to directly apply the knowledge gained and together implement and start their first pipeline with Nextflow. If you're interested in their feedback on the course, you can find it here.
Online Course - A Practical Introduction to NGS Data Analysis, March 2022
Posted on April 04, 2022
Our online course “A Practical Introduction to NGS Data Analysis” last week was a great event. The 22 participants from 12 different countries (Germany, Denmark, United States of America, United Kingdom, Sweden, Austria, Argentina, Netherlands, Slovakia, Belgium, Croatia, Hong Kong) were able to get a first insight into the world of NGS data analysis during the three days. By using our browser-based terminal, they were able to directly apply the knowledge gained and analyze their first data set. This will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level. If you're interested in their feedback on the course, you can find it here.
RNA-Seq Data Analysis Workshop, Feb. 2022 in Berlin
Posted on March 07, 2022

Our RNA-Seq Data Analysis Workshop in Berlin this week was a great event. Due to Covid-19, only vaccinated and recovered participants could take part (2G rule), but everyone felt safe and there was no mask requirement during the course. We were able to help the 12 participants perfectly because we were there with three trainers. The researchers got an introduction on how to work with linux and how to analyze RNA-Seq data correctly. The gained knowledge will allow them to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
ecSeq becomes partner of the future cluster SaxoCell®
Posted on February 21, 2022
Living Drugs - Precision therapy cluster for Saxony
SaxoCell® is one of the winners of the innovation competition "Clusters4Future" of the Federal Ministry of Education and Research (BMBF).
The creation of the future cluster is based on an association of leading research institutes and medical facilities from Saxony, including Leipzig and Dresden.
Together with numerous other partners from academia and industry, they have set themselves the goal of opening up new areas of application and production methods for cell and gene therapeutics by means of twelve innovative research and development projects, among other things.
In addition, SaxoCell® will strengthen regional networking within the research area and technology transfer for the benefit of sustainable, local value creation.
ecSeq is part of the SaxoCell® project SaxoCellOmics. This project is addressing optimal and early monitoring of the development and production of gene and cell therapeutics. SaxoCellOmics pursues four strategic goals for the cluster: (1) the provision of efficient and harmonized processes for monitoring the delivery and tolerability of novel therapies, (2) the identification of mechanisms of action, new targets and resistances, (3) the development of improved quality criteria for the manufacturing process, and (4) the development of predictive biomarkers.
Find more information about the research project and ecSeq's involvement on the SaxoCellOmics website.
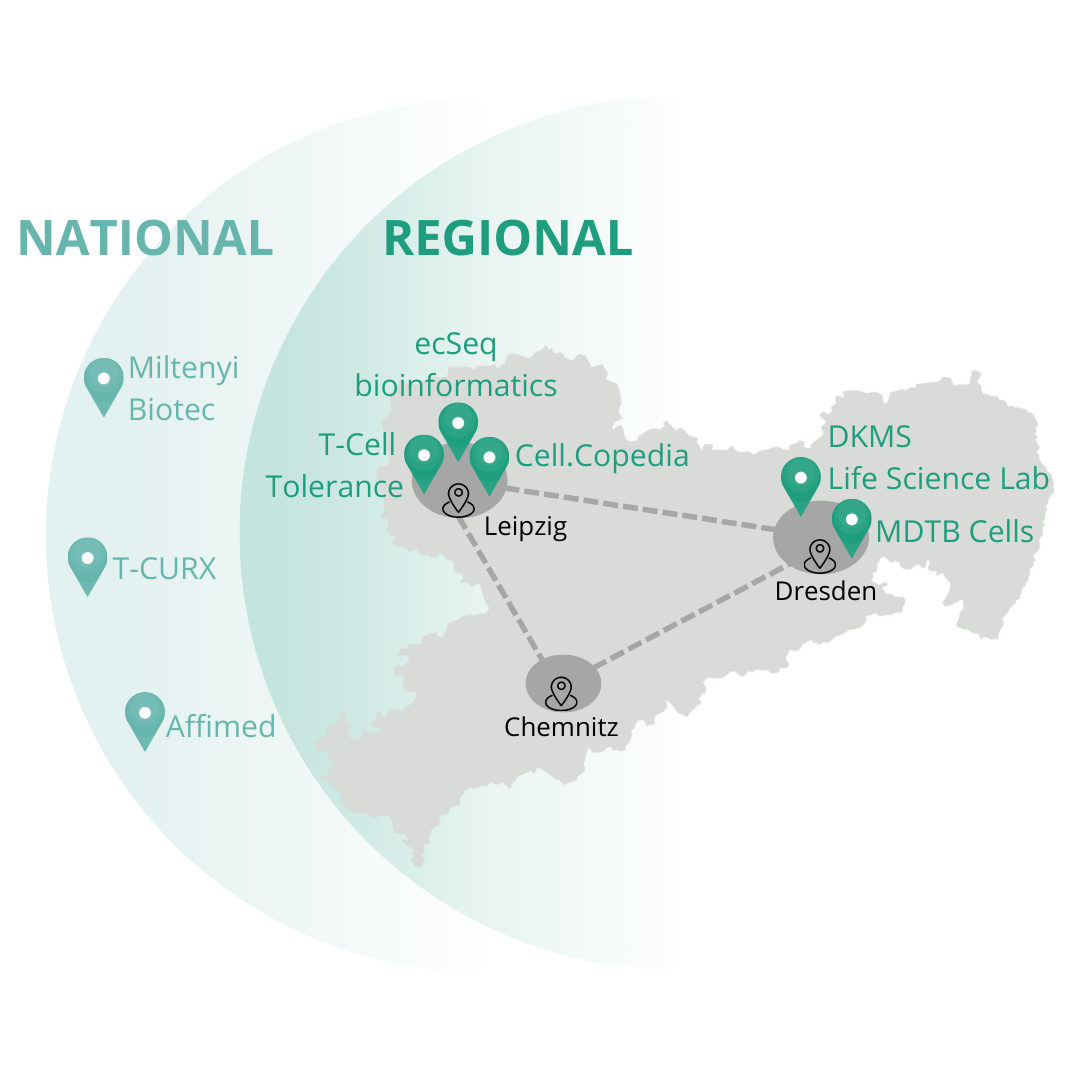
Online Course - A Practical Introduction to NGS Data Analysis, April 2021
Posted on December 13, 2021

Our online course “A Practical Introduction to NGS Data Analysis” last week was a great event despite the Covid-19 pandemic. The 30 participants from 12 different countries (Saudi Arabia, UK, Serbia, Germany, Romania, USA, Poland, Austria, Hong Kong, France, Switzerland, Croatia) were able to get a first insight into the world of NGS data analysis during the three days. By using our browser-based terminal, they were able to directly apply the knowledge gained and analyze their first data set. This will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level. If you're interested in their feedback on the course, you can find it here.
Online Course - Bioinformatics Pipeline Development with Nextflow, November 2021
Posted on November 26, 2021
Our online course “Bioinformatics Pipeline Development with Nextflow” this week was a great event despite the Covid-19 pandemic. The 18 participants from 9 different countries (USA, France, Hong Kong, Saudi Arabia, Belgium, Bulgaria, Netherlands, Denmark and Germany) learned how workflow management systems can accelerate their research. By using our browser-based terminal, they were able to directly apply the knowledge gained and together implement and start their first pipeline with Nextflow. If you're interested in their feedback on our courses, you can find it here.
Novel microRNA Biomarker Signature in Extracellular Vesicles for Atherosclerosis Detection
Posted on October 12, 2021
In cooperation with Prof. Dr. Gustav Schelling's group at the hospital of the Ludwig-Maximilians-University (LMU) Munich, Prof. Dr. Michael Pfaffl's group at the Technical University of Munich (TUM) and the company TIB MOLBIOL, we detected a new microRNA biomarker signature for risk assessment of arteriosclerosis and its sequelae. The first results are now published in Frontiers in Cell and Developmental Biology. This project was funded by ZIM, Germany's largest innovation programme for small and medium-sized enterprises. 'ZIM' stands for 'Zentrales Innovationsprogramm Mittelstand', which means 'Central Innovation Programme for small and medium-sized enterprises (SMEs)'. It is a funding programme of the Federal Ministry for Economic Affairs and Energy (BMWi) that aims to foster the innovative capacity of SMEs.
Reference:
Hildebrandt A, Kirchner B, Meidert A, Brandes F, Lindemann A, Doose G, Doege A, Weidenhagen R, Reithmair M, Schelling G, Pfaffl M: 'Detection of Atherosclerosis by Small RNA-Sequencing Analysis of Extracellular Vesicle Enriched Serum Samples', Front. Cell Dev. Biol. 10.3389/fcell.2021.729061 (2021)
Berlin Summer School in NGS Data Analysis, October 2021
Posted on October 09, 2021

Our 5th Berlin Summer School in NGS Data Analysis in Berlin this week was a full success. Due to Covid-19, this time only vaccinated and recovered participants could take part (2G rule), but everyone felt safe and there was no mask requirement during the course. We were able to help the 17 participants perfectly because we were there with three trainers. The researchers got an introduction on how to work with linux and how to analyze DNA-Seq and RNA-Seq data correctly. The gained knowledge will allow them to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
NGS Liquid Biopsy with Genetic Testing Platform 'Seamless NGS'
Posted on September 13, 2021

"Lung cancer is among the most frequently diagnosed cancers worldwide. Despite the availability of multiple targeted therapies, these tumors readily develop resistance mutations. To avoid taking invasive biopsies, these mutations can also be detected in liquid biopsies, i.e. blood samples derived from the patient. However, a reliable detection system such as NGS is required and in particular the bioinformatic analysis must be capable of detecting very low frequency variants. Recently, we successfully passed another QuIP round-robin test for the detection of EGFR resistance mutations in tissue as well as liquid biopsies using the Seamless NGS software"
Ulrike Obeck and Dr. Mathias Stiller (Institute of Pathology, Leipzig University Hospital)
Visit our website for the Seamless NGS Software Platform or download the brochure.

Improving the Sensitivity of Gene Fusion Detection from Archer® FusionPlex® Panels
Posted on August 13, 2021

Recent studies point to promising new therapeutic options that can be applied in the presence of certain gene fusion events (such as NTRK gene fusions). Diagnostic labs can use RNA sequencing to detect such fusion events. The source material (e.g. tumor tissue) has to be prepared for sequencing using custom-made assays or using commercial assays such as Archer® FusionPlex®.
Given that we recently introduced a new module for detecting fusion events to our Seamless NGS software platform, we were interested in how sensitive our approach is on existing data. But how is it possible to assess the detection level for fusion events? Ideally, there should be an experiment where the fusion events are contained in varying, but previously known, concentrations. One elegant solution: a "dilution series" type of experiment where a solution with the element to study is diluted in another solution at different concentrations.
This is exactly what Dilon et al. did in their publication "Targeted RNA-sequencing for the quantification of measurable residual disease in acute myeloid leukemia". Amongst other things, they diluted ME-1 cell line samples into healthy adult donor peripheral blood mononuclear cells with dilution ratios from 1:10 to 1:100,000. The ME-1 samples contain a known CBFB-MYH11 gene fusion.
Analysis Results
We obtained the raw RNA sequencing data which the authors have shared on a public sequence database. We focused on the ME-1 dilution series that was prepared with the Archer® FusionPlex® Myeloid assay and sequenced with an Illumina miSeq machine.
We then used the new Seamless NGS RNA Fusion workflow to detect fusion events. The algorithm is not limited to known fusion partners or to specific breakpoint locations, enabling the discovery of entirely new fusions. For each fusion event, detailed information is provided that can be used to filter for custom requirements. Besides basic measurements like read support and the fusions probability of being a driver of the oncogenic process, the software provides information about whether or not the fusion is found in databases like TCGA, ChiTaRS. Also, known diseases associations and relevant literature is included. In addition, each fusion event is visualized connecting the gene annotation with the read coverage and the detected breakpoints:
How to measure the abundance of a fusion? The original publication used the “percentage of unique reads spanning the breakpoint and supporting the event divided by the total reads at that locus” as a measure. Since “locus” was not clearly defined, we used a similar metric of “percentage of unique reads spanning the breakpoint and supporting the fusion event divided by the total number of reads at the breakpoint of the CBFB gene”.
The following plot shows these obtained percentages, first as shown in the publication (as computed by the original authors using the Archer® analysis software) and second with Seamless NGS.
| Cell Dilution | Orginal Study CBFB-MYH11 | Seamless NGS CBFB-MYH11 |
|---|---|---|
| 0 | 41.2% | 21.54% |
| 1:101 | 26.0% | 10.91% |
| 1:102 | 5.1% | 2.04% |
| 1:103 | 0.6% | 0.17% |
| 1:104 | Not detected | 0.01% |
Using the software provided with the Archer® assay, they were able to detect the CBFB-MYH11 fusion from dilutions of 1:10 to 1:1,000. However, our approach was able to detect the fusion in dilutions from 1:10 to 1:10,000. This means a 10-fold increased limit of detection for this fusion event.
An alternative way of measuring the abundance of a fusion is to normalize the support against a ubiquitously expressed gene, allowing a comparison between different samples and fusions. The authors are plotting the ratio of target copies/ABL1 copies (log10) against the cell dilution (−log10). They do this both for their custom assay (NIH AML MRD RNA-Seq) (blue) and the Archer® FusionPlex® Myeloid assay (red).
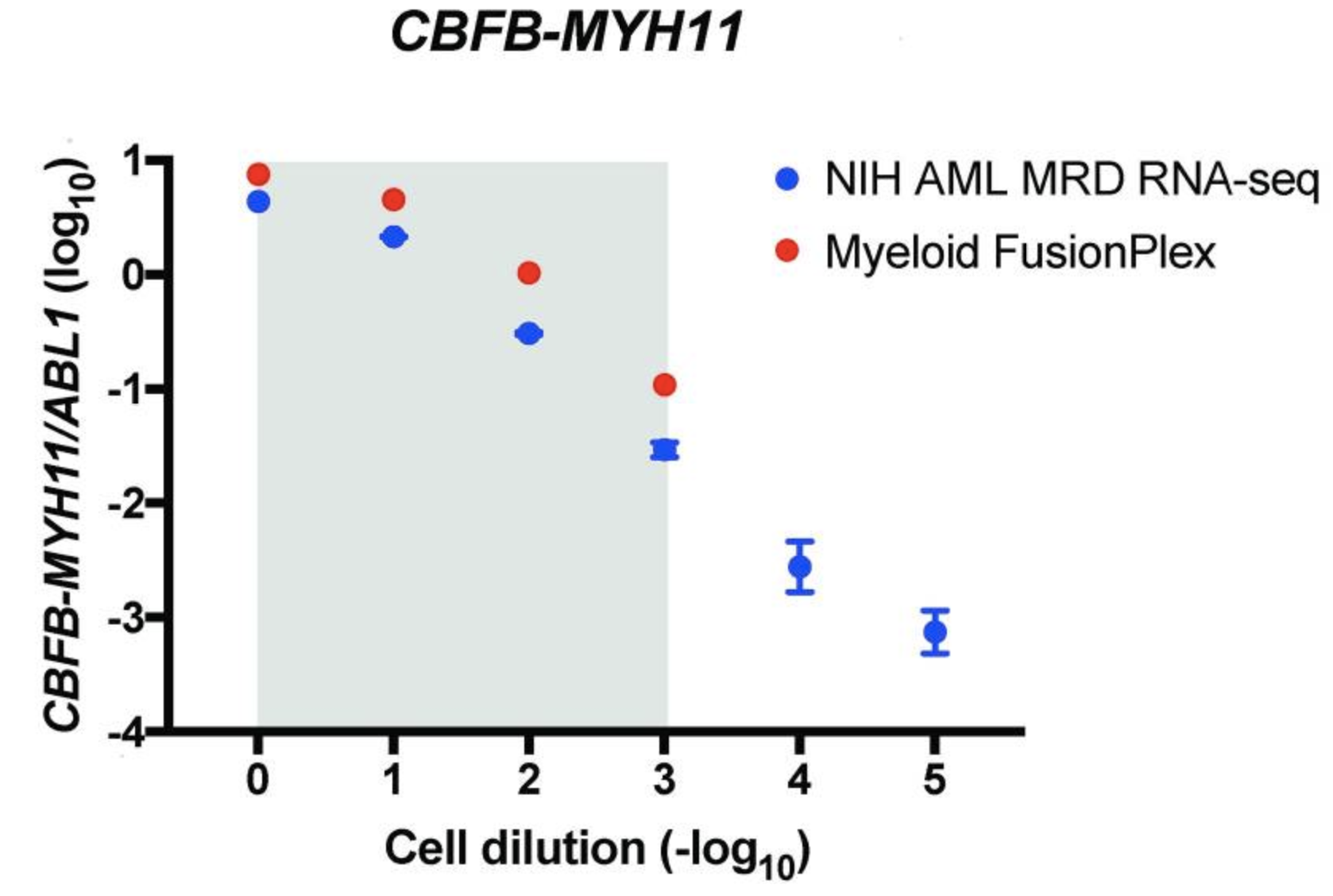
The grey area indicates the cell dilution frequencies in which the mutation could be detected by both methodologies. Note that dilution level 1:105 was not assessed with the FusionPlex assay. When we visualize the results of Seamless NGS similarly, we obtain the following plot:
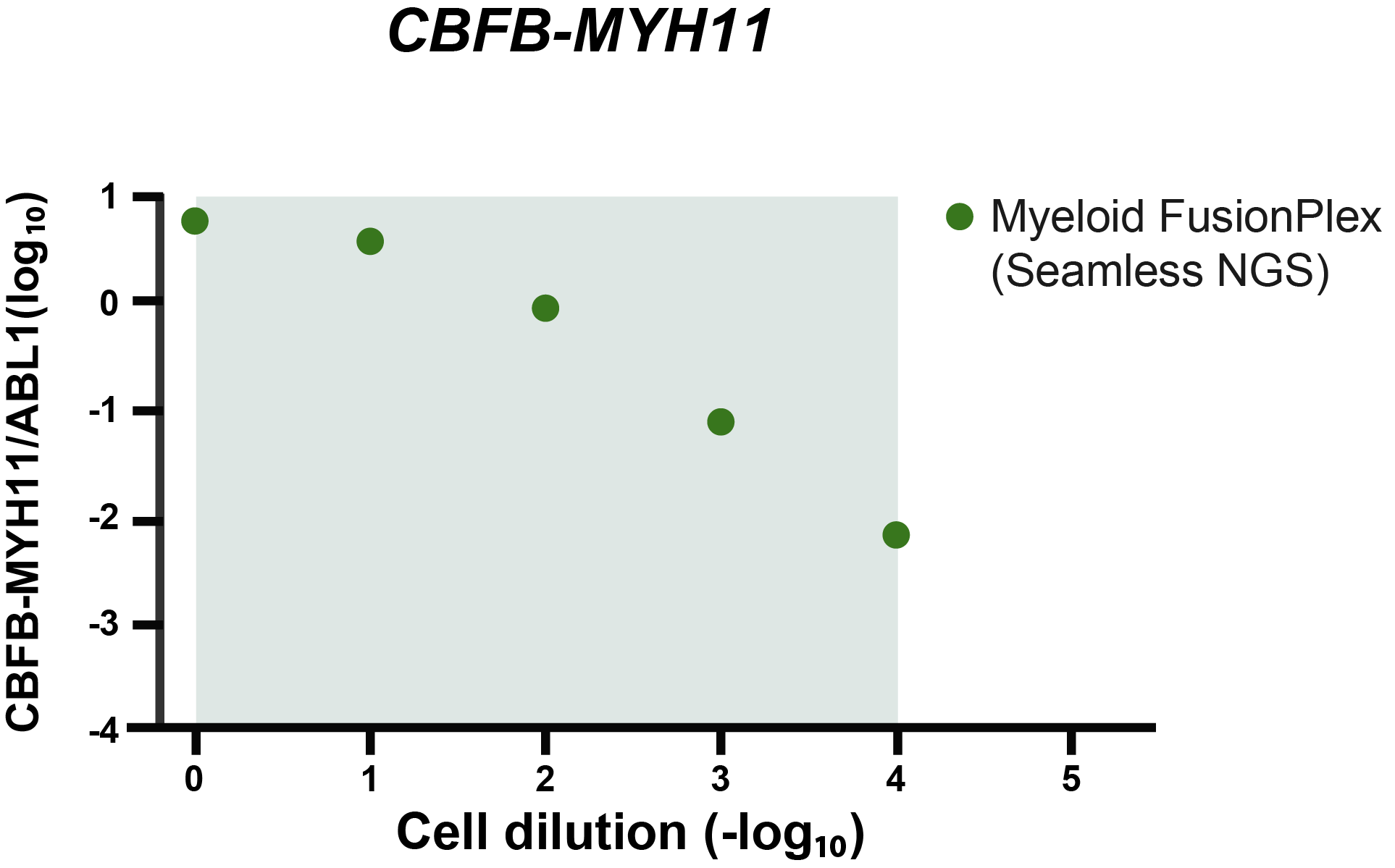
Again, we see that the abundance of fusion is linearly decreasing with the known dilution of the ME-1 cell lines. As indicated by the gray area, Seamless NGS can detect the CBFB-MYH11 fusion in the Archer® FusionPlex® Myeloid data down to a dilution of 1:104, whereas previous analysis methods from the same data allowed only detection levels of 1:103.
Summary
Fusion analysis with RNA sequencing offers great flexibility and sensitivity for detecting multiple molecular targets in a single assay. Comparing our results with previous results, we were happy to see that Seamless NGS did not only match the sensitivity of the Archer® software but surpasses it by one order of magnitude. This opens up new possibilities for users who would like a software that can flexibly analyze a range of RNA fusion assays from different vendors (Archer®, QiaSeq®, Illumina®, …) without sacrificing accuracy or convenience.
About Seamless NGS

Seamless NGS is a software platform for automated analysis and management of next-generation sequencing experiments. It provides easy access to advanced bioinformatics protocols for the detection of SNVs, InDels, Copy Number Variants and RNA Fusions. Seamless covers the complete genetic testing workflow from raw NGS data analysis to interpretation. Its private computing platform leaves any genetic customer data in-house, permitting GDPR compliance and full data control.
ecSeq at Week of the Environment in the park of Bellevue Palace with German President Frank-Walter Steinmeier
Posted on July 06, 2021

ecSeq at the "Week of the Environment" in the park of Bellevue Palace with German President Frank-Walter Steinmeier
The demand for tires for passenger cars, commercial vehicles, etc., but also for other rubber products, is causing a worldwide increase in demand for natural rubber. A truck tire, for example, contains up to 25 kg of the renewable rubber, whose excellent technical properties are currently not matched by synthetic polymers. To date, natural rubber has been obtained exclusively from the latex of the rubber tree Hevea brasiliensis, which grows in the tropics. In order to meet global demand in the medium term without tropical virgin forests having to make way for additional rubber plantations, alternative sources of natural rubber are needed. Russian dandelion Taraxacum koksaghyz, which can also be grown in temperate climates, produces a qualitatively comparable milky sap. In perspective, it is capable of diversifying rubber production and, in the process, preventing the depletion of endangered rainforests. However, Russian dandelion also offers the opportunity to increase the diversity of crops in domestic fields.
Find more information about the research project and ecSeq's involvement in this information leaflet by the FNR, the central coordinating agency in the area of renewable resources in Germany:

Automate your Twist Human Core Exome Kit Data Analysis with the Seamless NGS Platform
Posted on July 04, 2021

By using the Twist Bioscience Human Core Exome Kit together with the "Twist Human Core Exome" workflow of Seamless NGS you get a fully reproducible end-to-end analysis pipeline, from sample generation to annotated DNA variants (SNVs) and copy number variations (CNVs).
Your specialist can filter and examine relevant DNA mutations enriched with information from over 12 databases. The automatically generated report can then be downloaded or directly passed to your in-house LIMS system.
"Twist Bioscience NGS Target Enrichment Solutions offer superior enrichment efficiency. Modular kits for library preparation and target enrichment, as well as an optimized enrichment workflow and easy customization of panel content facilitate fast adoption and great results." Twist Bioscience
The Seamless NGS platform is optimized for fast and automated routine usage in diagnostic labs. The end-to-end software moves you rapidly from data generation to interpretation. In the 'Variant Explorer' you will get comprehensive information about DNA variants, their annotation and effects. Various sorting and filtering options will enable fast result interpretation.
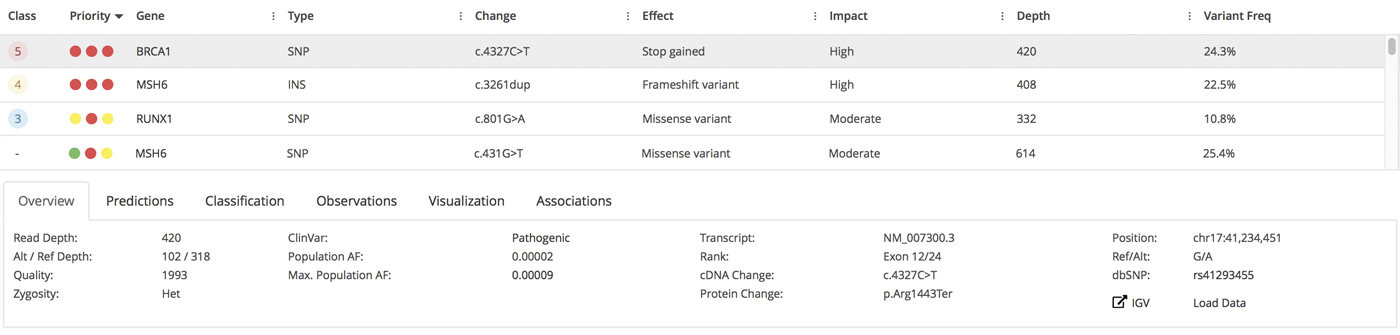
Online Course - Bioinformatics Pipeline Development with Nextflow, May 2021
Posted on May 07, 2021
Our online course “Bioinformatics Pipeline Development with Nextflow” this week was a great event despite the Covid-19 pandemic. The 14 participants from 9 different countries (USA, Singapore, France, Poland, Italy, UK, Czech Republic, Norway and Germany) learned how workflow management systems can accelerate their research. By using our browser-based terminal, they were able to directly apply the knowledge gained and together implement and start their first pipeline with Nextflow. If you're interested in their feedback on the course, you can find it here.
Online Course - A Practical Introduction to NGS Data Analysis, April 2021
Posted on April 30, 2021
Our online course “A Practical Introduction to NGS Data Analysis” this week was a great event despite the Covid-19 pandemic. The 24 participants from 9 different countries (Germany, Netherlands, Australia, Croatia, Spain, Canada, Poland, UK, USA) were able to get a first insight into the world of NGS data analysis during the three days. By using our browser-based terminal, they were able to directly apply the knowledge gained and analyze their first data set. This will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level. If you're interested in their feedback on the course, you can find it here.
Communicate Knowledge - New Article in Briefings in Bioinformatics
Posted on April 07, 2021

Whole genome bisulfite sequencing is currently at the forefront of epigenetic analysis, facilitating the nucleotide-level resolution of 5-methylcytosine (5mC) on a genome-wide scale. Specialized software have been developed to accommodate the unique difficulties in aligning such sequencing reads to a given reference, building on the knowledge acquired from model organisms such as human, or Arabidopsis thaliana. As the field of epigenetics expands its purview to non-model plant species, new challenges arise which bring into question the suitability of previously established tools. Herein, nine short-read aligners are evaluated: Bismark, BS-Seeker2, BSMAP, BWA-meth, ERNE-BS5, GEM3, GSNAP, Last and segemehl. Precision-recall of simulated alignments, in comparison to real sequencing data obtained from three natural accessions, reveals on-balance that BWA-meth and BSMAP are able to make the best use of the data during mapping. The influence of difficult-to-map regions, characterized by deviations in sequencing depth over repeat annotations, is evaluated in terms of the mean absolute deviation of the resulting methylation calls in comparison to a realistic methylome. Downstream methylation analysis is responsive to the handling of multi-mapping reads relative to mapping quality (MAPQ), and potentially susceptible to bias arising from the increased sequence complexity of densely methylated reads.
Reference:
Nunn A, Otto C, Stadler PF, Langenberger D: 'Comprehensive benchmarking of software for mapping whole genome bisulfite data: from read alignment to DNA methylation analysis', Briefings in Bioinformatics bbab021, https://doi.org/10.1093/bib/bbab021 (2021)

Adam Nunn holds a BSc in Biological Sciences from University of Portsmouth and a double MSc in Bioinformatics and Biology from Lund University, Sweden. Between the bachelor's and the master's degree, he worked for four years as a lead research scientist for a UK-based company where he focused on developing biological alternatives to chemical pesticides. In 2017 he joined ecSeq Bioinformatics GmbH for his PhD studies. His work is fully funded by the Marie Skłodowska-Curie Innovative Training Network EpiDiverse and in cooperation with Prof. Peter F. Stadler, holder of the bioinformatics chair at Leipzig University.
Online Course - A Practical Introduction to NGS Data Analysis, March 2021
Posted on March 05, 2021
Our online course “A Practical Introduction to NGS Data Analysis” this week was a great event despite the Covid-19 pandemic. The 22 participants from 9 different countries (Sweden, Slovenia, Norway, Australia, Germany, Switzerland, Canada, Denmark, Czech Republic) were able to get a first insight into the world of NGS data analysis during the three days. By using our browser-based terminal, they were able to directly apply the knowledge gained and analyze their first data set. This will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level. If you're interested in their feedback on the course, you can find it here.
5th Berlin Summer School - Meet Our Invited Speakers
Posted on February 22, 2021
It is our pleasure that, despite the Covid-19 pandemic, we are able to offer our summer school again this year.
We have a large room where the distances can be kept, there is an air filter in the room that removes the viruses from the air, we have scheduled regular breaks for ventilation and the hygiene concept of the room provider (walkways, disinfectants, mandatory masks in the hallways, etc.) has convinced us.
And the best for the participants: We guarantee that you will not lose your money if we are not allowed to hold the course because of Covid-19! You will get it back directly, or you can participate at an alternative date.
And now to the event! We would like to briefly introduce you to our Invited Speakers:

Meet our invited speaker Prof. Dr. Claude Becker
Dr. Becker is leader of the group 'Genetics and genomics of plant-environment interactions' and professor for Genetics at the Ludwig-Maximilians-University Munich.
Brief Resume
Dr. Becker studied biology in Heidelberg and received his diploma in 2005. Then, in 2010, he received his doctorate in biology from the University of Freiburg. He then joined the group of Dr. Detlef Weigel at the Max Planck Institute for Developmental Biology in Tübingen as a PostDoc. In 2016, he obtained his first own research group at the Gregor Mendel Institute in Vienna. Since 2021, he is professor for Genetics at the Ludwig-Maximilians-University Munich.
His research group is interested biochemical interactions in which plants produce and release chemical compounds to interfere with the growth or the development of their neighbors. Furthermore, his group studies the role of epigenetic marks in plant-environment interactions.

Meet our invited speaker Dr. Kevin Gesson
Dr. Gesson is Head of Research and Development at ViennaLab Diagnostics GmbH, Vienna, Austria.
Brief Resume
Dr. Gesson studied Biotechnology in Vienna at the University of Natural Resources and Life Sciences, Vienna (BOKU). As PhD student he went to the Max F. Perutz Laboratories in Vienna and received his doctoral degree in Biochemistry and Molecular Biology in 2016 with highest honors (summa cum laude) from the Medical University of Vienna.
He has been employed at ViennaLab Diagnostics GmbH since 2017 and is now leading the R&D department.

Meet our invited speaker Dr. Alexander Donath
Dr. Donath is head of the Computational Genomics section at the Zoological Research Museum Alexander Koenig, a research museum of the Leibniz Association.
Brief Resume
Dr. Donath studied computer science with a special focus on bioinformatics at the University of Leipzig and received his diploma in 2007. Between 2007 and 2011, he worked at the bioinformatics group of the University of Leipzig on his PhD thesis. He received his doctorate in 2011 from the University of Leipzig. Since 2011, he is staff scientist at the Zoological Research Museum Alexander Koenig in Bonn.
He specialized in the field of genome evolution, analyzes transcriptomes and genomes to answer phylogenetic questions, and coordinates the research facility's high-performance computing clusters.

Merry Christmas and a Happy New Year
Posted on December 25, 2020
As we look back on the past year, we would like to thank those who helped us navigate the difficult situation with the Corona Pandemic well. We enter 2021 with excitement and anticipation and hope that our success story will continue and that we will be able to carry out many exciting new projects.
Season’s greetings from the very merry team at ecSeq Bioinformatics.

Online Course - A Practical Introduction to NGS Data Analysis, December 2020
Posted on December 11, 2020
Our online course “A Practical Introduction to NGS Data Analysis” this week was a great event despite the Covid-19 pandemic. The 28 participants from 14 different countries (Germany, United Arab Emirates, Lithuania, Denmark, Australia, Hong Kong, Kuwait, Austria, Norway, Italy, UK, Netherlands, USA and Zimbabwe) were able to get a first insight into the world of NGS data analysis during the three days. By using our browser-based terminal, they were able to directly apply the knowledge gained and analyze their first data set. This will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Are Online Trainings for NGS Data Analysis as Effective as In-person Workshops?
Posted on November 06, 2020

Participating in a in-person workshop where everyone is in the same room and a trainer is present definitely has its advantages. Personal discussions and meetings are wonderful experiences. But especially now, during the Corona pandemic and the resulting restrictions, this is not always possible.
Now often the question arises 'Are online courses an alternative?'
The truth is that many of the participants are amazed at the value they get from our online courses.
Here are three powerful reasons to give it a try:
- Nothing is easier than attending an online course.
What could be easier than taking an online course? You can be on a different continent and participate in one of our courses online for several days. All you need is Internet access and a headset. - It is cost-effective.
With an online course you save time and money. You don't have to plan your travel and you don't have to worry about the logistics of accommodation. Likewise, you will not have any lost days of arrival and departure. - The quality is very high.
Our experts have created online learning plans that are as extensive as workshops that are held in person and offer plenty of opportunity for interaction. Same quality - just a different format.
What is special about ecSeq's online workshops?
We have invested a lot of time and effort to keep the differences between the online courses and the classroom courses as small as possible.
The theoretical part
Besides the typical webinar style where the trainer talks and shows things with the mouse on a shared slide, we also use traditional teaching methods. Here the trainer is filmed in front of the screen with the slides and can therefore teach more freely. Sometimes you simply need both hands to explain difficult procedures. If something needs to be drawn for an explanation, the screen of an iPad is shared, on which the trainer can write and draw properly. As you can see, we use all modern possibilities to get the most out of the online course. You will be amazed how effective this works.

The hands-on part
The biggest differences between our online courses and classroom courses can probably be found in the practical part. Here our developers have come up with something very special to make the experience of the practical part perfect. We give each participant access to a cloud computer on which they can perform the exercises. Using a common web browser, you can log on to this cloud computer and perform the exercises in a browser-based Linux terminal. You do not need to install anything for this.
And now comes the best:
Our trainers can look at your terminal in real time and even help you interactively. This is really like classroom training, where the trainers also walk through the rows and offer their help.
Note: The assistent can only see your in-browser terminal window but nothing else and also do not have any access to your computer.
The social interaction
This is probably the part where you understandably have the most disadvantages compared to a classroom course. You can't ask your table neighbor for help, you can't have a chat during the breaks and there are no networking events, such as the common dinner.
Nevertheless, we try to make the best out of the situation. In addition to a joint introduction round, in which everyone introduces themselves briefly, we use all possible communication channels to make the course as interactive as possible. Participants can raise their hands, unlock their microphones if they have questions, or communicate with each other in the chat. Each time we are surprised how much interaction is possible. Even short discussions between several participants are not unusual.
Summary
Of course, learning in an online course is different from learning in class. Nevertheless, during our first online courses we have noticed that the quality remains consistently high and is in no way less than that of a classroom course. The limitations of social interaction can be minimized by the technical possibilities. The financial and time advantages are and remain without question. The participants of our previous courses have given us only positive feedback, which shows us that online courses are a good replacement for classroom courses.
Nevertheless, we hope that the pandemic will soon come to an end and that we will again be able to face the participants of our courses directly. Until then, we hope that you all stay healthy!
Online Course - A Practical Introduction to NGS Data Analysis, October 2020
Posted on October 17, 2020
Our online course “A Practical Introduction to NGS Data Analysis” this week was a great event despite the Covid-19 pandemic. The 21 participants from 13 different countries (Lithuania, Belgium, Azerbaijan, Slovenia, Croatia, Latvia, Denmark, Germany, UK, China, Italy, Austria and Japan) were able to get a first insight into the world of NGS data analysis during the three days. By using our browser-based terminal, they were able to directly apply the knowledge gained and analyze their first data set. This will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Berlin Summer School in NGS Data Analysis, October 2020
Posted on October 12, 2020

Our 4th Berlin Summer School in NGS Data Analysis in Berlin last week was a full success despite the Covid-19 pandemic and the corresponding limitations and hygiene restrictions. The 14 participants had a perfect supervision key and were able to an introduction on how to work with linux and how to analyze DNA-Seq and RNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Nobel Prize in Chemistry awarded to Emmanuelle Charpentier
Posted on October 07, 2020

Huge congratulations to Emmanuelle Charpentier on the Nobel Prize
After we conducted an on-site course in the laboratory of Emmanuelle Charpentier (then at the Helmholtz Centre for Infection Research in Braunschweig) in 2014 to train her PhD students in the application of NGS data analysis, we have made our software Seamless NGS available to her research group. Emmanuelle is an extremely intelligent and determined woman who, despite her incredible success, is neither arrogant nor stuck-up. There are few successful scientists who have kept their feet on the ground and always talk to their conversation partner at eye level.
"I congratulate Emmanuelle on winning the Nobel Prize and wish her continued success for the future. Science needs exceptional people like her, who not only conduct outstanding research but are also able to communicate this to the outside world. Emmanuelle can do both. I am already looking forward to future projects with her." David Langenberger, CEO of ecSeq Bioinformatics GmbH
 © Nobel Media. Ill. Niklas Elmehed.
© Nobel Media. Ill. Niklas Elmehed.
RNA-Seq Data Analysis Workshop, September 2020
Posted on September 28, 2020

Our workshop “RNA-Seq Data Analysis - Quality Control, Read Mapping, Visualization and Downstream Analyses” in Leipzig last week was a full success despite the Covid-19 pandemic and the corresponding limitations and hygiene restrictions. The 9 participants had a perfect supervision key and were able to an introduction on how to work with linux and how to analyze RNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Save time and money through improved onboarding of your NGS data scientists
Posted on September 14, 2020

Hiring a suitable biologist or bioinformatician for sequencing data analysis takes lot of effort. Even if you are lucky and find the right talent, significant time is needed for on-the-job training. Your new team member needs time for learning before meaningful work results can be archived. This learning curve means that it could take weeks or even months for your new hiring to start working on his/her project.
The onboarding process is all about making employees, PhD students or Postdocs feel valued, providing them with the training and tools they need to succeed, and building a relationship with the workplace.
Employess with little or non onboarding often feel that they have to sink or swim alone, and are more likely to jump off the boat at the first opportunity. Since onboarding is the first real interaction of new team members with your company or institute, leveraging the experience is critical to avoid losing them.
We at ecSeq offer special NGS data analysis traingings, enabling your team members to quickly and purposefully enter this exciting field of work. With our training courses, the onboarding period can be reduced from several months to a few weeks. Starting with the basics (notations, methods, general understanding), we illustrate important error sources and slowly introduce participants to the Linux operating system. We show which tools can be used, how they have to be used and how one can interpret the results correctly. After our workshops, your new employees will be able to perform initial NGS data analyses independently and understand important technical NGS publications in detail.
Interested?
Several times a year, we organize introductory and advanced public courses on selected topics in next-generation sequencing data analysis.
Or do you want our trainers to come to you? Our on-site courses are very popular with companies or graduate schools from academic institutes or universities.
References:
We already trained many data scientists from large research organizations, universities, hospitals and large biotechnology and pharmaceutical companies:
- Bayer Crop Science
- KWS Saat AG
- F. Hoffmann-La Roche AG
- Miltenyi Biotec GmbH
- Max Planck Insitute for Biophysical Chemistry
- Max Planck Institute for the Science of Human History
- Max Planck Institute for Biology of Ageing
- Fraunhofer for Cell Therapy and Immunology
- Max Delbrück Center for Molecular Medicine
- Helmholtz Centre for Infection Research
- Robert Koch-Institute
- Weizmann Institute of Science
- Ludwig Maximilians University
- Charité Berlin
- and many more ...
Testimonials:
"Good resume of the latest technologies and applications. It definitely improves the communication with our bioinformatics colleagues to perfom the proper analysis and understanding of the data. Thanks for an intensive but nice course." Susana Gonzalez Fernandez-Nino, Bayer Crop Science
"Great Training! Competent trainers and enjoyed the practical part of the training very much." Joerg Schmiedle, F. Hoffmann-La Roche AG
"As a wet lab biologist, I have been dabbling in analysing my RNA-seq data myself. This course gave me a lot of context about mapping and differential expression software that I hadn't been able to grasp on my own. This knowledge will certainly improve the quality of my analyses. Additionally, I learned a lot of handy Linux commands and tricks that make my life a lot easier." Paul Essers, Max-Planck-Institute for the Biology of Ageing
"I was very positive surprised, how well the presenting and teaching of this Technology was. Overall the speed and the difficulty were very nice and helpful. The Organisation was well planned and the benefit that each of us, had a computer were very efficient for the direct excercise. So thanks a lot, I will definitely recommend this curse." Dominik Buob, CureVac AG
"The course was simply great! I was looking for an introduction to RNA seq analysis without need of prior experience and got exactly what I was looking for. The pace, the information and the structure of the course was perfect. It was a good mix of theory, going through exercises as a group and then trying it out for yourself to get a feel for it. I really learnt a lot, and the course leaders were extremely knowledgeable." Ida Jentoft, Max Planck Insitute for Biophysical Chemistry
"One of the best workshops I ever attended. The organization was just great. The small excercises during the workshop were quite useful to internalize the contents. It was fairly easy to follow even if one has not worked with linux before. I would strongly recommend the workshop!" Sophie Bartsch, Fraunhofer for Cell Therapy and Immunology
"Very valuable workshop which improved my bioinformatics understanding a lot and provided interesting aspects, I will put more focus on while analyzing our own data." Corinna S., KWS Saat AG
"I was amazed by the excellent course structure, preparedness of the teachers, and practical relevance. The course allows in immediate start in NGS RNAseq data analysis." Martin Sperfeld, Weizmann Institute of Science
"As someone who was previously terrified of the command line, this was a terrific course to build confidence in navigating the terminal. Moreover, I walked away with a much deeper understanding of sequencing chemistry and the relative merits of different software packages for analysing different types of sequencing data. I highly recommend this course for any wet lab biologist whether at the student, postdoc, or PI level!" Alexandra McCorkindale, Max Delbrück Center for Molecular Medicine
"Thanks for this great workshop. Even though I am no bioinformatician I learned a lot about the different steps of RNASeq data analysis. Not only the linux based commands but also the nice overview how the sequencing process itself works helped me a lot to get a deeper insight and understanding for library preparation and analysis." Lisa-Marie Schmid, Ludwig Maximilians University
Automate your Twist Human Core Exome Kit Data Analysis with the Seamless NGS Platform
Posted on September 13, 2020
By using the Twist Bioscience Human Core Exome Kit together with the "Twist Human Core Exome" workflow of Seamless NGS you get a fully reproducible end-to-end analysis pipeline, from sample generation to annotated DNA variants (SNVs) and copy number variations (CNVs).
Your specialist can filter and examine relevant DNA mutations enriched with information from over 12 databases. The automatically generated report can then be downloaded or directly passed to your in-house LIMS system.
"Twist Bioscience NGS Target Enrichment Solutions offer superior enrichment efficiency. Modular kits for library preparation and target enrichment, as well as an optimized enrichment workflow and easy customization of panel content facilitate fast adoption and great results." Twist Bioscience
The Seamless NGS platform is optimized for fast and automated routine usage in diagnostic labs. The end-to-end software moves you rapidly from data generation to interpretation. In the 'Variant Explorer' you will get comprehensive information about DNA variants, their annotation and effects. Various sorting and filtering options will enable fast result interpretation.
Whole-Exome Analysis on a Mainstream Desktop PC
Posted on September 12, 2020

Analysing NGS data usually is the domain of power compute servers or High-Performance Clusters, as it typically has massive volume and requires a lot of data crunching. However, the power of available CPUs is increasing, and now there are faster processors with more cores available on the market at competitive prices. This applies not only to the sector of the server “workhorses” but also to the sector of “standard” Desktop PCs. This sector is characterized by commodity hardware that is produced in high volumes and can be bought at affordable prices. While this kind of PC used to have just a single core, it is today possible to buy Desktop CPUs with as many as 16 cores.
Given the price premium that is necessary to enter the high-performance server sector, the question arises: why not use commodity hardware for NGS data analysis? So we started an experiment: how feasible is it to do a resource-intensive whole-exome sequencing (WES) analysis on a modern Desktop PC.
Hardware setup for WES analysis
We went on to assemble a suitable new computer with current hardware. We went for a 12-core AMD Ryzen 3900X CPU. This processor was released in 2019 and offers great performance. We then included 32 GB of RAM as well as a fast M2 SSD hard drive to make it suitable for the computation of NGS data. The total costs of this system were less than 1500 Euro.
We compare this to our 5-year old HPC unit that boasted an Intel Xeon processor with a total of 40 CPU cores. More CPU cores means that more computations can be conducted in parallel. The analysis will be much faster as long as the software is able to effectively make use of this parallelization. The HPC unit contained a RAID storage system with around 40 TB hard drives. At the time of buying this system had a price tag of around 12.000 Euro. So this is 800% the price of our new system.
As example data we took a full sequencing run of an Illumina NextSeq 500 that contained 16 whole-exome samples. Each was sequenced to an expected ~60x coverage using a Twist Bioscience Human Exome Kit with a target size of 36 Megabases. The sequencing run has a total sequencing depth of 100.4 Gigabases.
We analysed this data using the Seamless NGS software. Since our focus is to assess the performance gains due to hardware, not due to software, we used a (moderately optimized) standard analysis workflow relying on widely used NGS tools (e.g. BWA alignment). We previously confirmed that this approach for DNA variant detection meets our expectations regarding sensitivity (>0.99), specificity (>0.98) and reproducibility. Our analysis includes all auxiliary steps such as preprocessing, quality control and DNA variant annotation. Even though it is possible to perform Copy-Number Variation (CNV) analysis with Seamless NGS, it was excluded from the runtime analysis.
We were also interested whether there is a significant difference in the runtime between Linux and Windows, so we installed both operating systems on the new computer. Seamless NGS runs on both of these, so we could use that for comparison. For technical reasons we could not install Windows on our HPC.
What were our initial expectations before running the experiment? Our HPC unit is quite powerful. With 12 vs 40 CPU cores, it has more than 3 times as many physical processors available for challenging computations. On the other hand we have seen considerable improvements of computer hardware in the last years, including the availability of affordable SSD drives as well as the release of new processors with higher per-core performance. So we think that it is entirely possible that the NGS analysis performance of the new system is on par with our HPC unit.
WES runtime comparison results
Before looking at the performance of the different systems, we need to establish that we obtain comparable analysis results in all the test cases. To this end, we compare several important QC measures - the number of detected variants, alignment rate, mean coverage - across the different systems. We find that these numbers are absolutely identical - so we can go on.
Let us first look at the runtime on the High-Performance Cluster (on Linux). We will visualize the time required for the analysis separated by individual analysis steps. We here omit small steps that take only a few seconds and also group these steps into broader categories (e.g. Quality Control) for good oversight. Let’s take a look at the resulting Gantt chart:
The total runtime is about 21 hours - this is slightly below 2 hours per sample. This is not a bad time, considering that all necessary steps (including annotation) are already included in this analysis. Most of that time is spent with the alignment, variant calling and preprocessing raw reads.
Next, let's take a look at the runtime of the same analysis on our new 1500-Euro computer, running Linux:
Wow! The total runtime on this is about 10h, or about 50 minutes per sample. This result beat our wildest expectations. Yes, we expected that hardware improvements would lead to a strong improvement on a per-core basis. After all, hardware manufacturers are constantly innovating and competing hard over every performance gain. We did not expect that the 12-cores would in fact beat the 38-core HPC by THAT much. From this cursory analysis it is not entirely clear to us what factor(s) are responsible for these performance gains, but likely it will be a combination of increased CPU speeds, CPU processing efficiency, storage speed and mainboard bus throughput.
Now let’s take a look on what happens when we use the same machine, with the same data and analysis, but switch the operating system from Linux to Windows 10:
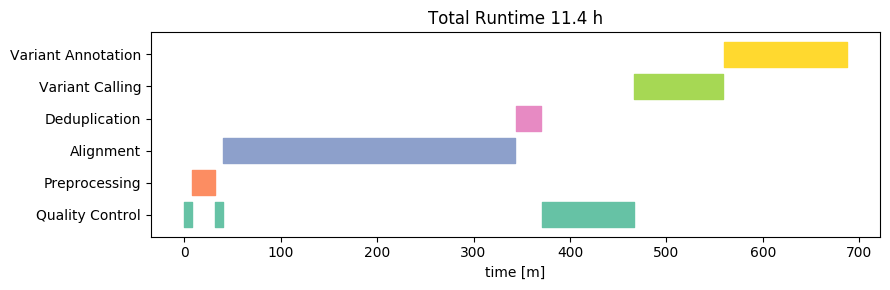
In this case, the runtime is slightly higher on Windows compared to that on Linux on the same machine. The difference however is not substantial - each sample takes about 55 minutes. Most of the difference takes place in the alignment step.
Finally, let us look at the total runtime differences:
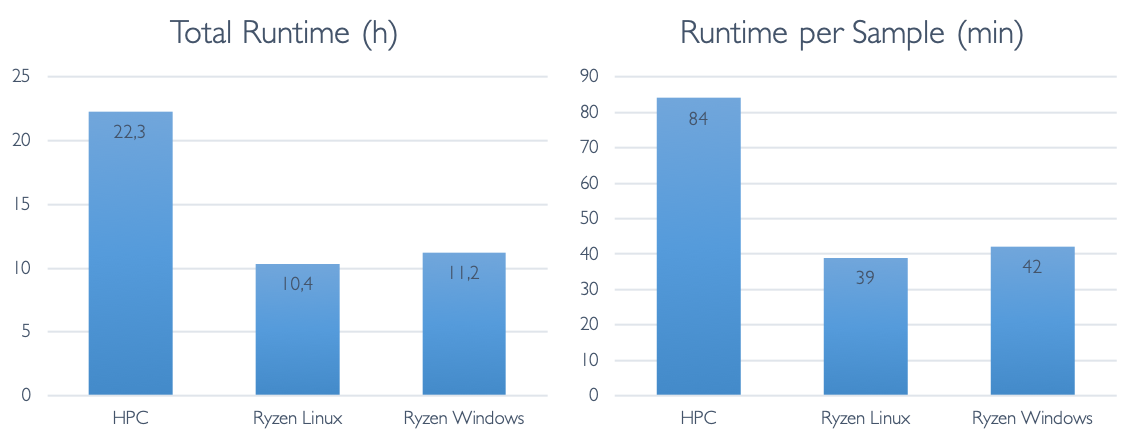
Outlook
Are there faster ways to analyse Whole-Exomes, Whole-Genomes or Clinical Exomes? Absolutely. You could replace standard tools with high-performant software more suited to the specific data. You could employ dedicated computer hardware such as Field Programmable Gate Arrays (FPGAs) or Graphical Processing Units (GPUs). In fact, it is possible to purchase commercial systems that combine some of these techniques to bring down the computation time to as low as 8 minutes per exome from reads to DNA variants. However, the goal of this post was a different one: to show how far you can go today without a big investment.
The results of our test really surprised us: it is possible to easily analyse two NextSeq 500 runs comprising 24 Whole Exomes in a single day on a ‘normal’ Desktop PC. This means that even such a low-cost system is able to analyse more than 8000 exomes per year! We expect that with a bit more optimization, significantly higher numbers will be possible - on a single machine. Most software tools will also allow you to combine several machines. Does this mean it is time to rethink the hardware requirements needed for NGS data analysis, including Whole-Exome Sequencing?
About Seamless NGS
Seamless NGS is a software platform for automated analysis and management of next-generation sequencing experiments. It provides easy access to advanced bioinformatics protocols for the detection of SNVs, InDels, Copy Number Variants and RNA Fusions. Seamless covers the complete genetic testing workflow from raw NGS data analysis to interpretation. Its private computing platform permits hassle-free GDPR compliance and full data control.
Transferring patient's genetic testing data to the cloud: 5 myths
Posted on September 09, 2020

We often get the question: how safe is it to transfer patient’s genomic data to the cloud? By this most people refer to the transfer of their NGS-based patient data to external web-based services (e.g. Illumina Basespace) that run on the public cloud of other third-party providers (e.g. Amazon AWS).
Of course, patient genetic data security should be a top priority. New sequencing technologies create large data volumes for each patient, and often small labs and hospitals assume that they cannot cope with the required increase in computational resources, leaving the cloud the only option. After all, cloud-based solutions are good for cases where varying amounts of data must be processed flexibly. But it is also true that a patient’s genetic testing data has a different privacy value compared to for example videos, music, images or other large data. Genetic testing data contains highly personal information, including health, behavioural and biometric information, and is an even more unique marker than a fingerprint. A DNA data breach is much worse than a credit card breach.

With that in mind, let us address the following misconceptions and myths we often hear.
Myth 1: "But I am using anonymous IDs instead of patient names”
A good practice in clinical diagnostics is the use of anonymized patient identifiers instead of real names. In the age of genomics, however, this is not sufficient anymore. Everybody working in genetics should internalize this: there is no effective way to anonymize genetic data! It is absolutely possible to infer the person from any sufficiently large genetic data, because the data is intrinsically identifying.
For this very reason, genetic data is considered Personally Identifiable Information (PII) according to Europe’s privacy law GDPR. Other PII includes fingerprints, real names, identification numbers, location data and online identifiers (e.g. IP addresses). In addition, because genetic data can reveal additional information about a person, such as ethnicity, it is explicitly part of “special categories of personal data” according to Article 9 of the GDPR. That means that the GDPR puts in tight regulations for the handling of genetic data itself. These include intense security requirements, limited data access guidelines and non-trivial obtaining of legally watertight consent. Any sloppiness with or even leakage of genetic data therefore comes not only with a loss of reputation but also potentially large GDPR-related compensations.
Myth 2: "But the IT security of vendor X surely is better than ours"

It might be true that some vendor X has a great IT security team and therefore is able to secure their servers very well. Yet, your data still is likely less secure. Consider that the total risk for data leakage is not the risk of the weakest part; it is the combined risk of all involved parties. Some IT specialists describe cloud as “just someone else’s computer”. In practice several parties (e.g. the web service provider, the infrastructure provider, third-party service providers) potentially have access to the computing and storage devices that contain patient data. Obviously, the more parties have access to data, the higher the “attack surface” which describes the potential for data leaks. This total risk should be minimized.
A leakage can occur at any involved party, be it your lab, a service vendor, additional cloud vendors etc. Consequently, you should be aware of and audit the security of all involved parties. There are vendors that perhaps you don’t even think of in the first place, such as the payment processing vendor that is linked to a 2019 breach of patient records of more than 19M american patients.
When a leakage occurs, it is usually difficult to determine who is really at fault, as can be seen in the case of AWS and Capital One. The legal consequences in such a case are difficult to assess and insurance companies will only compensate when it is clearly established who has committed a mistake. But, no matter who is at fault, the reputation of involved diagnostic labs will be ruined.
As a final point, if any malicious actor would be able to obtain access to your labs computer infrastructure, it is likely that they will be able to use this to access information actually available via external providers or services. So total risk should always include the local lab security.
Myth 3: "But the data is transferred encrypted, e.g. using a VPN"
In order for the data to be analysed at external vendors, they must have complete access to the raw, unencrypted data. Nobody can do meaningful data analyses with encrypted data. To display patient reports to you, those reports must be stored at the vendors database. When you use a VPN to work with the external service over the internet, the data is only encrypted on the way from your computer to the service provider. Actually, a similar kind of data “transport encryption” is activated on nearly every website you visit by default using SSL (see the lock symbol in your browser bar). This means that any additional communication security between your web browser and service provider is mainly window dressing, with no real security benefit.
Myth 4: "But our vendor is certified (e.g. HIPAA or medical device)"
There are many certifications that can be applied in the scope of genetic data processing. Many of them do not specifically address IT security, or do so only at a very basic level. Certification guidelines are often not able to cope with the speed of development, for example the massive increase in genomic data that is now possible with the newest generation of sequencing machines. As a case in point, it is known that the “HIPAA does not consider genome sequences as identifying information that has to be removed under the Safe Harbor Method for deidentification”. With the new data amounts, genomic data cannot be considered deidentifiable anymore. You are responsible for establishing appropriate security levels and performing due diligence rather than relying on maybe unrelated or obsolete certifications.
Myth 5: “But I need the scalability of the cloud for NGS analyses”
The major benefit of public clouds is that, in some minutes, you will be able to use dozens of computers that can do work for you. This is a highly appreciated feature, in particular for research making use of large data volumes over short analysis “peaks”. From our experience, most routine labs are able to handle large data volumes and sequencing throughout themselves. We have seen labs with dozens of sequencers that process their data in-house easily, and probably have significant cost savings with respect to cloud computing. Often internal computer infrastructure for workstations, LIMS, medical records servers are maintained anyway. Also, you would perhaps be surprised what a single server with optimized software can compute: 30 exomes in a day are doable.
The fingerprint analogy
Fingerprints are, like genetic data, personally identifying information. Nowadays many phones include a fingerprint sensor making access to your mobile phone both secure and convenient. For this feature, your phone must be able to store and analyse fingerprints. How does it secure your fingerprint from potentially malicious apps installed on your phone? Does it maybe even send you fingerprint for backup to the cloud?
It turns out, phone developers took these concerns seriously and worked out a secure way to deal with fingerprints. Android phones are required to use a special isolated area on the phone, called TEE, to store and analyse fingerprint data. This often is a separate CPU with its own memory and own operating system, completely isolated from the rest of the phone’s system. When you register a fingerprint on your phone, the scan from the sensor is sent to the TEE which creates validation data and encrypted fingerprint data. With this encryption, it is not possible to make sense of this data even if some app would manage to get hold of it. Hence there is no way that the fingerprint ever leaves the phone. All the validations are done in the TEE, and only cryptographic proofs of the validation are delivered.

Summary
A lot of misbeliefs can be heard in the area of genomic data security, in particular in the field of cloud services. Cloud computing brings convenience: customers don't need to install software and can outsource IT services and infrastructure. But there might be cases where risks, data privacy regulations and practical considerations outweigh this convenience. The sensitive information intrinsic to genetic data in the genomics age is often overlooked. If you would hesitate transferring customer fingerprints, patient names and medical reports to the cloud, then you should make no exception to modern genetic testing data.
This article is available in other languages: German version
NGS Data Analysis Workshop, July 2020
Posted on July 27, 2020

Our course “Next-Generation Sequencing Data Analysis: A Practical Introduction” in Munich last week was a great event despite the Covid-19 pandemic and the corresponding limitations and hygiene restrictions. The 8 participants had a perfect supervision key and were able to get a first insight into the world of NGS data analysis during the three days. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Improving the Sensitivity of Gene Fusion Detection from Archer® FusionPlex® Panels
Posted on March 19, 2020
Recent studies point to promising new therapeutic options that can be applied in the presence of certain gene fusion events (such as NTRK gene fusions). Diagnostic labs can use RNA sequencing to detect such fusion events. The source material (e.g. tumor tissue) has to be prepared for sequencing using custom-made assays or using commercial assays such as Archer® FusionPlex®.
Given that we recently introduced a new module for detecting fusion events to our Seamless NGS software platform, we were interested in how sensitive our approach is on existing data. But how is it possible to assess the detection level for fusion events? Ideally, there should be an experiment where the fusion events are contained in varying, but previously known, concentrations. One elegant solution: a "dilution series" type of experiment where a solution with the element to study is diluted in another solution at different concentrations.
This is exactly what Dilon et al. did in their publication "Targeted RNA-sequencing for the quantification of measurable residual disease in acute myeloid leukemia". Amongst other things, they diluted ME-1 cell line samples into healthy adult donor peripheral blood mononuclear cells with dilution ratios from 1:10 to 1:100,000. The ME-1 samples contain a known CBFB-MYH11 gene fusion.
Analysis Results
We obtained the raw RNA sequencing data which the authors have shared on a public sequence database. We focused on the ME-1 dilution series that was prepared with the Archer® FusionPlex® Myeloid assay and sequenced with an Illumina miSeq machine.
We then used the new Seamless NGS RNA Fusion workflow to detect fusion events. The algorithm is not limited to known fusion partners or to specific breakpoint locations, enabling the discovery of entirely new fusions. For each fusion event, detailed information is provided that can be used to filter for custom requirements. Besides basic measurements like read support and the fusions probability of being a driver of the oncogenic process, the software provides information about whether or not the fusion is found in databases like TCGA, ChiTaRS. Also, known diseases associations and relevant literature is included. In addition, each fusion event is visualized connecting the gene annotation with the read coverage and the detected breakpoints:
How to measure the abundance of a fusion? The original publication used the “percentage of unique reads spanning the breakpoint and supporting the event divided by the total reads at that locus” as a measure. Since “locus” was not clearly defined, we used a similar metric of “percentage of unique reads spanning the breakpoint and supporting the fusion event divided by the total number of reads at the breakpoint of the CBFB gene”.
The following plot shows these obtained percentages, first as shown in the publication (as computed by the original authors using the Archer® analysis software) and second with Seamless NGS.
| Cell Dilution | Orginal Study CBFB-MYH11 | Seamless NGS CBFB-MYH11 |
|---|---|---|
| 0 | 41.2% | 21.54% |
| 1:101 | 26.0% | 10.91% |
| 1:102 | 5.1% | 2.04% |
| 1:103 | 0.6% | 0.17% |
| 1:104 | Not detected | 0.01% |
Using the software provided with the Archer® assay, they were able to detect the CBFB-MYH11 fusion from dilutions of 1:10 to 1:1,000. However, our approach was able to detect the fusion in dilutions from 1:10 to 1:10,000. This means a 10-fold increased limit of detection for this fusion event.
An alternative way of measuring the abundance of a fusion is to normalize the support against a ubiquitously expressed gene, allowing a comparison between different samples and fusions. The authors are plotting the ratio of target copies/ABL1 copies (log10) against the cell dilution (−log10). They do this both for their custom assay (NIH AML MRD RNA-Seq) (blue) and the Archer® FusionPlex® Myeloid assay (red).
The grey area indicates the cell dilution frequencies in which the mutation could be detected by both methodologies. Note that dilution level 1:105 was not assessed with the FusionPlex assay. When we visualize the results of Seamless NGS similarly, we obtain the following plot:
Again, we see that the abundance of fusion is linearly decreasing with the known dilution of the ME-1 cell lines. As indicated by the gray area, Seamless NGS can detect the CBFB-MYH11 fusion in the Archer® FusionPlex® Myeloid data down to a dilution of 1:104, whereas previous analysis methods from the same data allowed only detection levels of 1:103.
Summary
Fusion analysis with RNA sequencing offers great flexibility and sensitivity for detecting multiple molecular targets in a single assay. Comparing our results with previous results, we were happy to see that Seamless NGS did not only match the sensitivity of the Archer® software but surpasses it by one order of magnitude. This opens up new possibilities for users who would like a software that can flexibly analyze a range of RNA fusion assays from different vendors (Archer®, QiaSeq®, Illumina®, …) without sacrificing accuracy or convenience.
About Seamless NGS
Seamless NGS is a software platform for automated analysis and management of next-generation sequencing experiments. It provides easy access to advanced bioinformatics protocols for the detection of SNVs, InDels, Copy Number Variants and RNA Fusions. Seamless covers the complete genetic testing workflow from raw NGS data analysis to interpretation. Its private computing platform leaves any genetic customer data in-house, permitting GDPR compliance and full data control.
4th Berlin Summer School - Meet Our Invited Speakers
Posted on March 17, 2020

Meet our invited speaker Prof. Dr. Manja Marz
Dr. Marz is leader of the group 'non-coding RNAS in aging: the regulation of aging' at the Leibniz Institute on Aging and full professor for 'High Throughput Sequencing Analysis' at Friedrich Schiller University Jena.
Brief Resume
Dr. Marz studied biology and computer sciences in Leipzig, Edinburgh and Darmstadt. She finished in 2005 her first diploma in biology and in 2006 her second diploma in computer science. Between 2006 and 2009 she worked on her PhD thesis in bioinformatics and got her doctorate degree in 2009 from the University of Leipzig.
In 2010 she moved to Marburg and started a Junior-Professorship in "High Throughput Sequencing Analysis" and became leader of a 'RNA Bioinformatics' group at Philipps-University Marburg. In 2015 she got a full profssorship for "High Throughput Sequencing Analysis" at Friedrich Schiller University Jena and a research group at the Leibniz Institute for Age Research – Fritz Lipmann Institute. She is also scientific advisory board member of RNA Central and Rfam, Founding member, board member and managing director of “European Virus Bioinforamtics Center” and Founding and board member of ZAJ (Aging Research Center Jena).

Meet our invited speaker Dr. Kristin Reiche
Dr. Reiche is leader of the Bioinformatics Unit at the Fraunhofer Institute for Cell Therapy and Immunology (IZI) in Leipzig.
Brief Resume
Dr. Reiche studied computer science and medical informatics in Leipzig and Liverpool. In 2003 she finished her diploma and started her PhD studies at the bioinformatics chair of Leipzig University. She got her doctoral degree in 2007 with highest honors (summa cum laude).
In 2011 she started a postdoc position at the RNomics group at the Fraunhofer Institute for Cell Therapy and Immunology (IZI) and in 2012 she moved to the Helmholtz-Centre for Environmental Research (UFZ) in a research group for bioinformatics and transcriptomics. In 2012 she spent three month at the Broad Institute of the MIT and Harvard in Cambridge, USA. Since 2015 she is head of the bioinformatics unit at the Fraunhofer Institute for Cell Therapy and Immunology (IZI) and since 2019 she is a senior research scientist at the Institute of Clinical Immunology of the Medical Faculty at Leipzig University.

Meet our invited speaker Niroshan Nadarajah
Mr. Nadarajah is head of Bioinformatics Innovation & Partner Management at the Munich Leukemia Laboratory (MLL).
Brief Resume
Niroshan Nadarajah studied computer science with a special focus on bioinformatics at Heinrich Heine University Düsseldorf and received his M.Sc. in 2011. After researching at a bioinformatics group at UC Davis in California (US) and working as a senior web developer for a German media agency, he started working for the Munich Leukemia Laboratory (MLL) in 2011. Here, he started the development of a software pipeline for NGS diagnostics. He developed a web application to manage quality control and interpretation of NGS/laboratory processes. After some years as a team leader of 6 bioinformaticians, he is now head of Bioinformatics Innovation. He introduces advanced methods and technologies into routine diagnostics workflows, including Cloud computing for NGS and AI methods for phenotype analysis for cytomophology and immunophentyping. In 2017 and 2018 he was member of the MLL management board.

RNA-Seq Data Analysis Workshop, March 2020
Posted on March 06, 2020

Our workshop “RNA-Seq Data Analysis - Quality Control, Read Mapping, Visualization and Downstream Analyses” in Berlin this week was a full success. Our two trainers gave the 29 participants from 14 different countries (UK, Germany, Netherlands, France, Poland, Belgium, United Arabic Emirates, Ireland, USA, Denmark, Switzerland, Czech Republic, Australia, Norway) an introduction on how to work with linux and how to analyze RNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Follow us on social media
Posted on February 14, 2020

No? Feel free to follow us!

On Twitter we do not only share promotions about our services, but also overall news about NGS. What new methods are there on the market? What will be of interest in the future? Follow us and stay up-to-date!
Follow ecSeq on Twitter

On LinkedIn we have both, a company page on which we share news and updates, as well as three discussion groups. In the groups, a broad audience of experts shares their own news to keep everyone updated. All members and messages in the groups are specific to the groups topic.
Follow ecSeq on LinkedIn
Group: NGS Data Analysis
Group: Diagnostic Laboratories
Group: microRNA Research

On facebook we have a company page and a very active group for NGS data analysis. People from all over the world ask questions, discuss and inform the followers about everything related to NGS and bioinformatics.
Follow ecSeq on Facebook
Group: NGS Data Analysis

For our German followers we have a company page and a discussion group for beginners in NGS data analysis in Xing.
Follow ecSeq on Xing
Group: NGS Data Analysis
Rubber from russian dandelions
Posted on January 16, 2020
Joint project with Fraunhofer IME
Russian dandelion (Taraxacum koksaghyz) produces a rubber of comparable quality to the tropical rubber tree. It also grows in temperate climates and is thus able to expand natural rubber production to our regions. The cultivation of dandelion also offers the opportunity to increase the diversity of cultivation on domestic fields.
Russian dandelion is a close relative of our native dandelion. It enriches in its roots a whitish, sticky, rubbery milky sap, the so-called latex. Since dandelions are only of limited use as a wild plant for agricultural production, plant breeders clarify the physiology of rubber synthesis and develop suitable varieties for successful industrialization. These should be characterized by high root yields, latex content and resistance to plant diseases, pests and drought. And, like sugarbeet, the roots must be easy to clear and as clean as possible and then easy to store and process.
To achieve this, together with the Fraunhofer Institute for Molecular Biology and Applied Ecology IME, ESKUSA GmbH and hortilab, ecSeq Bioinformatics GmbH generated a high-resolution genetic map of Russian dandelion.
This is used to achieve the following breeding objectives:
- Stabilize yields and, in the long term, increase them to approximately one ton of natural rubber per hectare per year
- Optimise root geometry to minimise soil attachment, contamination and losses during harvest
- Improve drought tolerance
- Increase resistance to disease
Find more information about the research project and ecSeq's involvement in this information leaflet by the FNR, the central coordinating agency in the area of renewable resources in Germany:
Merry Christmas and a Happy New Year
Posted on December 23, 2019
As we look back upon the past year, we would like to acknowledge those who have helped us shape our business. Thanks for a great year, and we wish you all the best as you embark on 2020.
Season’s greetings from the very merry team at ecSeq Bioinformatics.

Annual Workshop Review 2019
Posted on December 18, 2019

We say THANK YOU
The NGS introduction workshop for beginners last week was the last for this year. Thus, we want to thank all participants for their attendance, their thirst for knowledge and their great questions. It was a pleasure for us to give you first impressions of NGS, some insights into the main issues when working with this sequencing method and a starting point for your own analyses.
In our eight public workshops this year we had more than 150 participants from 37 different countries.
 Map with the countries of origin of all participants
Map with the countries of origin of all participants
The participants of this years workshops came from Austria, Belgium, Brazil, Bulgaria, China, Croatia, Czech Republic, Denmark, Estonia, Finland, France, Germany, Guatemala, Hungary, Ireland, Israel, Italy, Lithuania, Mexico, Netherlands, Norway, Poland, Portugal, Republic of Korea, Romania, Russia, Saudi Arabia, Slovenia, Spain, Sweden, Switzerland, Thailand, UK, United Arabic Emirates and USA.
What courses are planned for 2020?
For 2020 we will have seven public workshops in Leipzig, Berlin and Munich. If you like to join, please check our upcoming courses in 2020.
Some impressions from 2019








NGS Data Analysis Workshop, December 2019
Posted on December 16, 2019

Our workshop “Next-Generation Sequencing Data Analysis: A Practical Introduction” in Munich this week was a full success. Our two trainers gave the 25 participants from 12 different countries (Switzerland, Austria, UK, Germany, Lithuania, Czech Republic, Italy, Spain, Poland, USA, Saudi Arabia and France) an introduction on how to work with linux and how to analyze RNA-Seq and DNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
NGS Workshop Schedule for 2020
Posted on November 18, 2019

Finally, our workshop schedule for 2020 is online. Based on the availibility of our trainers and extensive feedback from former participants and interested persons, we put together the following courses and dates.
NOTE: Since the Berlin Summer Schools fill up pretty fast every year, you should apply fast to catch your seat.
UPCOMING NGS DATA ANALYSIS WORKSHOPS IN 2020:
RNA-Seq Data Analysis Workshop
- Date: March 2-5, 2020
- Location: Berlin, Germany
Next-Generation Sequencing Data Analysis: A Practical Introduction
- Date: March 25 - 27, 2020
- Location: Munich, Germany
4th Berlin Summer School 2020
- Date: June 15-19, 2020
- Location: Berlin, Germany
RNA-Seq Data Analysis Workshop
- Date: September 21-24, 2020
- Location: Leipzig, Germany
Next-Generation Sequencing Data Analysis: A Practical Introduction
- Date: October 14-16, 2020
- Location: Munich, Germany
DNA Methylation Data Analysis
- Date: November 2-5, 2020
- Location: Berlin, Germany
Next-Generation Sequencing Data Analysis: A Practical Introduction
- Date: December 9-11, 2020
- Location: Munich, Germany
DNA Methylation Data Analysis Workshop, November 2019 in Berlin
Posted on November 18, 2019
Our workshop “DNA Methylation Data Analysis” in Berlin last week was a full success. Our two trainers gave the 7 participants from 7 different countries (Finland, Sweden, Czech Republic, Bulgaria, Saudi Arabia, Austria and Belgium) an introduction on how to work with linux and how to analyze Bisulfite-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
NGS Data Analysis Workshop, October 2019
Posted on October 28, 2019

Our workshop “Next-Generation Sequencing Data Analysis: A Practical Introduction” in Munich this week was a full success. Our two trainers gave the 15 participants from 12 different countries (Ireland, Poland, Romania, UK, Denmark, Norway, Austria, Switzerland, Portugal, Italy, Sweden and Germany) an introduction on how to work with linux and how to analyze RNA-Seq and DNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Learning NGS Data Analysis in Great Cities
Posted on October 25, 2019
Upcoming events in 2019
There are four more workshops this year. Get trained in NGS Data Analysis and enjoy one of three great German cities in summer, fall or winter.
Believe us, every city is worth a trip.
Bisulfite-Seq in Berlin, Germany
Berlin in winter: Discover the city for yourself in your very own way. The Brandenburg Gate with its glittering icicles, the Tiergarten under a wonderful white blanket of snow - winter in Berlin is simply wonderful.

DNA Methylation Data Analysis
How to use bisulfite-treated sequencing to study DNA methylation
November 12-15 in Berlin
In a nutshell
- Learn how bisulfite sequencing works
- Understand how bisulfite-treated reads are mapped to a reference genome
- Perform basic analyses (call methylated regions, perform basic downstream analyses)
- Use shell scripting to create reusable data pipelines
- Visualize results (ready-to-publish)
The purpose of this workshop is to get a deeper understanding of the use of bisulfite-treated DNA in order to analyze the epigenetic layer of DNA methylation. Advantages and disadvantages of the so-called 'bisulfite sequencing' and its implications on data analyses will be covered. The participants will be trained to understand bisulfite-treated NGS data, to detect potential problems/errors and finally to implement their own pipelines. After this course they will be able to analyze DNA methylation and create ready-to-publish graphics.
NGS-Intro in Munich, Germany
English Garden, Nymphenburg Castle, Isar: Munich is beautiful in winter. Swim outside in the steaming Westbad in Pasing and watch the surfers on the Eisbach wave in the English Garden. And in the evening, visit one of the many Christmas markets and enjoy a hot mulled wine.

NGS Data Analysis - A Practical Introduction
Quality Control, Read Mapping, Visualization and DNA Variant Analysis
December 11-13 in Munich
In a nutshell
- Learn the essential computing skills for NGS bioinformatics
- Understand NGS analysis algorithms (e.g. read alignment) and data formats
- Use bioinformatics tools for handling RNA-Seq data
- Perform first downstream analyses for studying genetic variation
The purpose of this workshop is to get a deeper understanding in Next-Generation Sequencing (NGS) with a special focus on bioinformatics issues. Advantages and disadvantages of current sequencing technologies and their implications on data analysis will be discovered. The participants will be trained on understanding their own NGS data, finding potential problems/errors therein and finally perform their first downstream analysis (variant calling). In the course we will use a real-life NGS dataset from the current market leader illumina.
RNA-Seq Data Analysis Workshop, September 2019
Posted on September 27, 2019

Our workshop “RNA-Seq Data Analysis - Quality Control, Read Mapping, Visualization and Downstream Analyses” in Leipzig this week was a full success. Our two trainers gave the 23 participants from 11 different countries (Germany, France, Austria, Belgium, Finland, UK, Estonia, Norway, Israel, Denmark, Sweden) an introduction on how to work with linux and how to analyze RNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Dr. Langenberger on analyzing Qiagen's QiaSeq DNA Panels using Seamless NGS Software
Posted on August 19, 2019
Commercial library preparation kits for NGS instruments are getting increasingly complex in order to obtain high sensitivity while reducing bias and errors immanent in current sequencing technology. One approach offered by Qiagen Inc. in their QiaSeq Targeted DNA Panels is using a combination of single primer extension and molecular barcodes.
In March, Dr. David Langenberger, CEO of ecSeq Bioinformatics, chaired the 'NGS & NGS Data Analysis' session at the '9th Gene Quantification Event qPCR dPCR & NGS 2019'. He also held a talk with the title "Tiled Amplicons Panels in NGS-Based Genetic Testing" where he spoke about the challenges involved analyzing such panels. An active discussion was carried about the question whether anyone but experiences NGS bioinformaticians will be able to use standard software or build in-house pipelines for complex panels. Dr. Langenberger also presented Seamless NGS as push-button software solution with excellent support for QiaSeq Targeted Panels.
For more information on the software solution please visit Seamless NGS Website or download the brochure.
Here you can have a look at the presentation.

Privacy implications of genetic data sharing
Posted on July 08, 2019
Data is the new oil has been the slogan of recent years, explaining the tremendous rise in power and the data hunger of tech companies such as Facebook. Emails, contacts, social interactions, online activity, payments, hobbies, interests, location profiles - everything is collected. However there are increasing concerns with the current practices of handling these vast treasures of data, particularly with third-party sharing, its exploitation for unethical purposes and leakage of not sufficiently secured data. These practices leave customers with uncomfortable feelings and spurn initiatives advocating for more privacy and security regulations.
Meanwhile, consumer genetic testing companies are advertising a range of low-priced products offering personalised health or ancestry information from nothing more than a bit of your saliva. Many are wondering: wouldn't one of those kits be a good present for friends, relatives or themselves? Working in the Bio-IT field, I feel that many are underestimating the amount of highly personal information that can be gained from genetic testing, and are therefore overlooking the consequences of sharing their genetic data with these companies. With that in mind, let's take a look at what genetic information can reveal about you.
Genetic data as unique human identifier
Due to the imperfections of biology, no two humans have identical DNA. Even identical twins that start with the same DNA from a single fertilized egg will accumulate mutations over time. Some occur so early in embryonic development that the majority of cells will inherit them, leading to small but detectable differences in their genetic code (1). DNA therefore is a durable and unique marker of human identity, and much more precise than other biometric markers such as fingerprints.
Genetic data can reveal your identity using databases
There exist various databases collecting genomics information from consumer genetic tests. As shown in a 2018 study your genetic data can be used to reveal your identity with impressive accuracy. As of the time of the study, it was estimated that 60% of people with European descendancy can be pinpointed to a third-cousin or closer match based on existing data. With more and more genomic data becoming available, these numbers can grow rapidly.
 Figure 1: A hypothetical family tree showing the reach of public genetic databases. In this example, one of your third cousins, who shares the same great-grandparent as you, posts their test results and makes you identifiable. Figure by Aparna Nathan (article)
Figure 1: A hypothetical family tree showing the reach of public genetic databases. In this example, one of your third cousins, who shares the same great-grandparent as you, posts their test results and makes you identifiable. Figure by Aparna Nathan (article)
Already, these techniques are being increasingly utilised by law enforcement. In May 2019, big discussions were sparked by a case of violent assault where DNA was uploaded to a database for comparison with available genetic data, leading to the identification of a suspect. Critics fear that this technique will be increasingly used for less serious crimes, particularly as there is astonishingly little oversight regarding your own right to genetic privacy if you are identified in this manner. The success of this technique shows: your genetic data can never be anonymous.
Genetic data provides phenotypic information
DNA phenotyping is the task of predicting a person’s physical features using only genetic data. This a very active research area. It is already possible to determine eye color, hair color and skin color with high confidence. A 2017 study successfully used genomic data to predict facial structure, voice, biological age, height, weight and other human physical traits. There are already companies that commercially offer to predict the physical appearance of an unknown person from only their DNA, as obtained for example from a tiny bit of saliva on a cigarette butt.
 Figure 2: Actual (left) and predicted from DNA (right) faces (from this study).
Figure 2: Actual (left) and predicted from DNA (right) faces (from this study).
When asked, people usually underestimate how much of these physical traits are due to genetic factors versus environment. Heritability is the scientific term describing how much of a trait, for example differences in body height, can be attributed solely to genetic differences. The heritability of eye color for instance is 95%, so eye color could nearly completely be determined by your genes. The heritability for body weight (!) and height is 80% and 70%, respectively. Consequently, it can be expected that advances in DNA phenotyping will enable determination of more and more accurate, biometric information from genetic data in the near future.
Genetic data contains behavioural information
A long-standing question, not only among behavioural scientists, is how much of our personal character is determined by genetics and how much is due to our environment (e.g. education). There are an increasing number of studies suggesting that the majority of psychological differences can be attributed to genetic factors. As psychologist Robert Plomin writes in his 2018 book “Blueprint”:
In summary, parents matter, schools matter, and life experiences matter, but they don’t make a difference in shaping who we are. DNA is the only thing that makes a substantial systemic difference, accounting for 50 percent of the variance in psychological traits. The rest comes down to chance environmental experiences that do not have long-term effects.
Some of the traits that were already examined for heritability include school achievement (60%), verbal ability (60%), spatial ability (70%) and ability to remember faces (60%). All of these attributes are encoded in our genetic data. As with phenotypic information, it is therefore increasingly possible to predict a person's behavioural traits based on only DNA.
Genetic data provides health information
Some hereditary diseases such as cystic fibrosis, sickle cell disease, and haemophilia are related to abnormalities in a single gene, and can be identified by reading out your DNA. Many other disorders are related to multiple genes in combination with environmental factors. These are known as complex disorders.
A relatively new development is the possibility to determine an individual's risk for common complex disorders like coronary artery disease, atrial fibrillation and type-2 diabetes. So-called polygenic risk scores now make it possible to compute a significant fraction of increased risk for these diseases. This is done by combining multiple genetic markers into a single score to predict lifetime outcomes. For example a polygenic risk score could be established for the onset of the severe mental illness schizophrenia (80% heritability). These scores can be helpful information for doctors when considering treatment options. However, information about disabilities or mental health should deserve the highest protection. Think about how employers, insurance or finance institutions could predicate their actions if it were known that an onset of schizophrenia is likely. As of today, no company who has genetic data is obliged to tell you about the findings and judgements from it.
Genetic data contains information about your relatives as well
Following the laws of inheritance your parents, siblings and kids share roughly half of their genetic information with you. Other relatives also share significant parts: you share 12.5% of DNA with your cousin, and about 0.8% with a third-cousin. Therefore sharing your genetic data always means sharing information about your relatives. It means that it cannot be avoided that genetics testing companies are getting more information than they imply, and you should always consider the consequences for your relatives’ interests and rights.
Awareness for genetic privacy
It cannot be overstated: intensely private and personal information - in a surprising breadth and depth - can be gained from genetic data. It could be summarised like this in the oversimplified formula:
Genetic data = fingerprint + passport + appearance + behaviour + health information
So who shall you trust with the handling of your genetic data? We have become more and more accustomed to events of "we are sorry that we have to report a cybersecurity incident", from even the most technologically potent firms. So is it worth it to demand higher security standards for genetic testing companies? Given the power of genetic data, I think higher security requirements are a must. Leaking of material information is bad, but at least you can react. You can change passwords, emails, credit card numbers, in emergency even addresses. But you cannot change your genetic code.
In June 2019 it became known that personal, financial and medical data of the two largest american genetic testing companies, LabCorp and Quest Diagnostics, had been leaked, totalling to 20M patients. Both companies shared this data with a third-party provider which had been breached. It is a simple truth: the more parties with access to sensitive data, the more likely eventual data leakage becomes. Furthermore, consumer genetic testing companies tend to have a second business as data brokers, selling your genetic data to third parties.
It is a valid demand for consumers and patients to have a right for genetic data privacy and security. The GDPR explicitly includes “genetic information” in their data privacy regulation, which is a step in the right direction, but greater awareness about what we are giving away with this kind of data is of utmost pertinence in this fast-growing market. Not every company has data security as their top priority, so consumers need to maintain their own due diligence to make sure there is sufficient trust when sharing the deep information inherent in genetic data.
Footnotes:
(1) Consumer genetic tests will typically read out only the highly-variable sites (polymorphisms) instead of the complete DNA. For anyone except identical twins these tests will give significant differences. Some information might only be extracted from more comprehensive “genomic” data. But due to the growing possibilities for imputation, i.e. increasing the information from limited available data using the data from others, the partial genetic data from consumer genetic tests is often sufficient.
Bringing the power of Big Data to biomedical researchers
Posted on June 28, 2019
Great article about ecSeq Bioinformatics's work and vision in the 'Healthcare Journal of Baton Rouge'. Thanks to Dr. Claudia S. Copeland (LinkedIn Profile) for this understandable article about a very complex topic

Read Flipping Book Version
Read PDF Version
Conversations with Bioinnovators - Dr. Christian Otto
Posted on June 26, 2019

Some weeks ago, our head of software development Christian Otto was interviewed by Dr. Claudia S. Copeland from Carpe Diem Biomedical Writing for the 'Conversations with Bioinnovators series'. Christian talked about his work as our lead software developer and achieving work-life balance.
Read the full interview here:
"Dr. Christian Otto on NGS-based research, empowering medical professionals through user-friendly software, and achieving work-life balance"
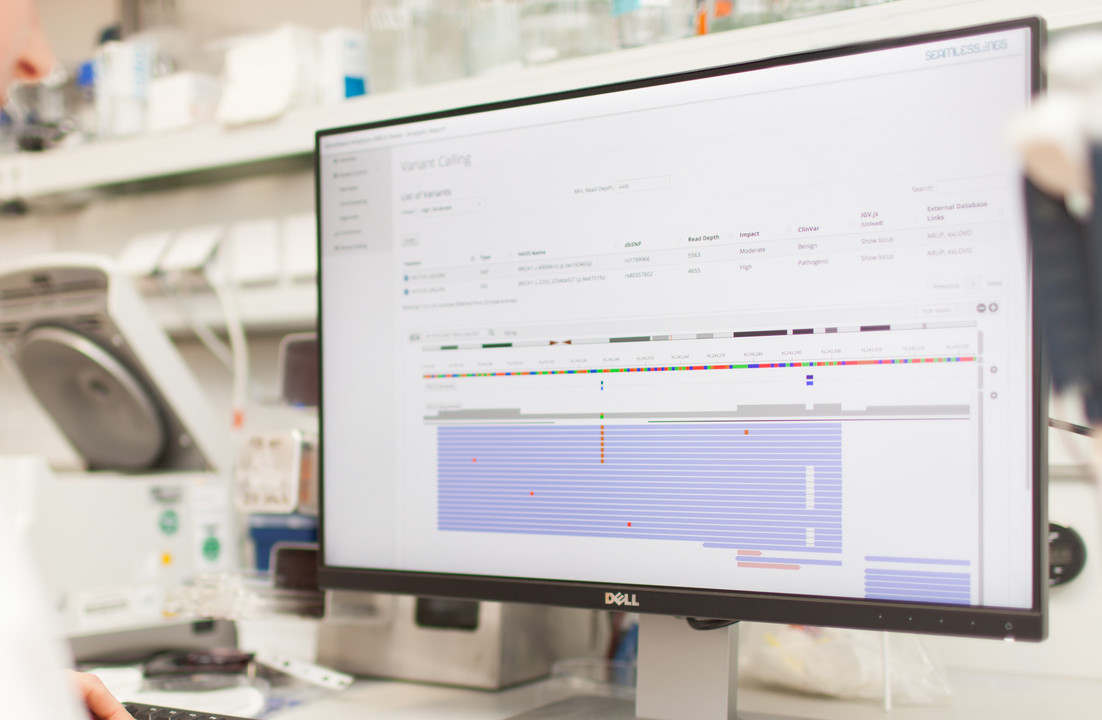
Diagnostic test for risk assessment of arteriosclerosis
Posted on June 24, 2019
R&D cooperation project - Diagnostic tests for risk assessment of arteriosclerosis and its sequelae
Arteriosclerosis is probably the most common disease worldwide. With increasing age, the number of patients with significant arteriosclerotic changes increases, and it can be assumed that practically every patient over 80 years of age has at least one form of this vascular disease. The age distribution in the western world will lead to a further increase in arteriosclerotic systemic diseases. In Germany, arteriosclerosis and its complications led to 3500 days of inpatient hospitalisation per 100,000 inhabitants in 2015.
Typical secondary diseases of arteriosclerosis with considerable epidemiological and economic significance are myocardial infarction, stroke, aortic aneurysms caused by arteriosclerosis, peripheral arterial occlusive disease and chronic renal insufficiency.
Arteriosclerosis as a cause of cardiovascular disease is currently responsible for most deaths and illnesses in industrial societies. If the diagnosis is made in good time, arteriosclerosis can be treated (with medication, change of behaviour) and the majority of its sequelae can be prevented (especially myocardial infarction or renal insufficiency). Today, however, a diagnosis is usually made too late and only when acute symptoms and damage have already occurred. Treatment can then usually only alleviate symptoms, but in many cases no complete cure can be achieved.
The aim of the project is the development of a 'liquid-biopsy' based diagnostic test procedure, which for the first time will allow an individual assessment of the risk of arteriosclerosis and its organ-specific sequelae.
This project will be in cooperation with the hospital of the Ludwig-Maximilians-University (LMU) Munich, the Technical University of Munich (TUM) and the company TIB MOLBIOL and is funded by ZIM, Germany's largest innovation programme for small and medium-sized enterprises. 'ZIM' stands for 'Zentrales Innovationsprogramm Mittelstand', which means 'Central Innovation Programme for small and medium-sized enterprises (SMEs)'. It is a funding programme of the Federal Ministry for Economic Affairs and Energy (BMWi) that aims to foster the innovative capacity of SMEs.
3rd Berlin Summer School, June 2019
Posted on June 21, 2019

Our 3rd Berlin Summer School in NGS Data Analysis in Berlin this week was a full success. Our three trainers gave the 26 participants from 15 different countries (Thailand, Germany, USA, Poland, France, Denmark, Norway, Brazil, Republic of Korea, Israel, Saudi Arabia, Russia, Switzerland, Sweden, Croatia) an introduction on how to work with linux and how to analyze DNA-Seq/RNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.









103rd Annual Meeting of the German Society of Pathology
Posted on May 27, 2019
 ecSeq will participate the 103rd Annual Meeting of the German Society of Pathology (DGPatho). The event will take place in Frankfurt am Main from June 13th to 15th. Stop by and check our genetic testing software Seamless NGS. We are excited to giving first impressions of our new features and having fruitful discussions with interested users.
ecSeq will participate the 103rd Annual Meeting of the German Society of Pathology (DGPatho). The event will take place in Frankfurt am Main from June 13th to 15th. Stop by and check our genetic testing software Seamless NGS. We are excited to giving first impressions of our new features and having fruitful discussions with interested users.
If you attend this event, feel free to visit our booth (B10, 2nd floor) and chat with our team. They will answer all your questions and give you some first impressions on how Seamless NGS can help you with your routine genetic analyses.
We're looking forward to meet you in Frankfurt am Main.
NGS Data Analysis Workshop, May 2019
Posted on May 15, 2019

Our workshop “Next-Generation Sequencing Data Analysis: A Practical Introduction” in Munich this week was a full success. Our two trainers gave the 28 participants from 17 different countries (Hungary, Croatia, Belgium, China, Slovenia, Guatemala, Denmark, Germany, Netherlands, UK, Italy, USA, Russia, Brazil, Spain, Thailand and Ireland) an introduction on how to work with linux and how to analyze RNA-Seq and DNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
EpiDiverse - Infering Genomic Information from WGBS Data
Posted on April 18, 2019
Our Phd student Adam Nunn explains how to infer genomic information from Bisulfite-Seq data
EpiDiverse - Doing a Doctorate at ecSeq
Posted on April 17, 2019
Adam Nunn is Doing his Doctorate at ecSeq
Adam holds a BSc in Biological Sciences from University of Portsmouth and a double MSc in Bioinformatics and Biology from Lund University, Sweden. Between the bachelor's and the master's degree, he worked for four years as a lead research scientist for a UK-based company where he focused on developing biological alternatives to chemical pesticides. In 2017 he joined ecSeq Bioinformatics GmbH for his PhD studies. His work is fully funded by the Marie Skłodowska-Curie Innovative Training Network EpiDiverse and in cooperation with Prof. Peter F. Stadler, holder of the bioinformatics chair at Leipzig University.

![]()
Linking Ecology, Molecular Biology and Bioinformatics in plant epigenetic research
EpiDiverse is a Marie Skłodowska-Curie Innovative Training Network aimed at the study of epigenetic variation in wild plant species. The network joins research groups from ecology, molecular (epi)genetics and bioinformatics to explore the genomic basis, molecular mechanisms and ecological significance of epigenetic variation in natural plant populations. This project received funding from the EU Horizon 2020 program under Marie Skłodowska-Curie grant agreement No 764965 ... read more
Outstanding research - New Article in Nature
Posted on April 05, 2019

Burkitt lymphoma is the most common B-cell lymphoma in children. Within the International Cancer Genome Consortium (ICGC), Dr. Gero Doose, our head of research and development, performed transcriptome sequencing data analysis of 39 sporadic Burkitt lymphoma. Dr. Doose and colleagues unraveles the interaction of structural, mutational, and transcriptional changes, which contribute to MYC oncogene dysregulation together with the pathognomonic IG-MYC translocation.
Reference:
López C, Kleinheinz K, Aukema SM, Rohde M, Bernhart SH, Hübschmann D, Wagener R, Toprak UH, Raimondi F, Kreuz M, Waszak SM, Huang Z, Sieverling L, Paramasivam N, Seufert J, Sungalee S, Russell RB, Bausinger J, Kretzmer H, Ammerpohl O, Bergmann AK, Binder H, Borkhardt A, Brors B, Claviez A, Doose G, Feuerbach L, Haake A, Hansmann ML, Hoell J, Hummel M, Korbel JO, Lawerenz C, Lenze D, Radlwimmer B, Richter J, Rosenstiel P, Rosenwald A, Schilhabel MB, Stein H, Stilgenbauer S, Stadler PF, Szczepanowski M, Weniger MA, Zapatka M, Eils R, Lichter P, Loeffler M, Möller P, Trümper L, Klapper W; ICGC MMML-Seq Consortium, Hoffmann S, Küppers R, Burkhardt B, Schlesner M, Siebert R.: 'Genomic and transcriptomic changes complement each other in the pathogenesis of sporadic Burkitt lymphoma.'. Nature Communications, 10.1038/s41467-019-08578-3 (2019)
Dr. Gero Doose is head of research and development at ecSeq Bioinformatics GmbH. He developed RNA folding and protein solubility prediction algorithms before he started working with high-throughput sequencing data. Gero implemented detection pipelines for circularized and trans-spliced transcripts. As an expert for split-read alignment analysis, he also developed a sophisticated method to detect differential alternative splicing.
EpiDiverse - 2nd Annual Meeting
Posted on April 01, 2019

ecSeq welcomed the complete EpiDiverse consortium in Leipzig to review the progress of the ITN project
From Thursday 28th March 2019 to Friday 29th March 2019, all EpiDiverse beneficiaries came together for the 2nd annual meeting at Leipzig University. The 15 Early Stage Researchers (ESRs) presented their research and spent a complete day to discuss the progress of the EpiDiverse projects.



As a beneficiary of EpiDiverse, ecSeq supervises one ESR, provides NGS trainings to all ESRs and operates the EpiDiverse high-performance computer clusters.
3rd Berlin Summer School - Meet Dr. Giovanni Marco Dall'Olio
Posted on March 25, 2019

Meet our invited speaker Dr. Giovanni Marco Dall'Olio
Dr. Giovanni Marco Dall'Olio is a bionformatics investigator at Glaxo Smith Kline (GSK) in Stevenage, UK.
We are very happy to have Dr. Giovanni Marco Dall'Olio at our Summer School. He made the transition from academia to big pharma and can tell about pitfalls and opportunities.
Brief Resume
Dr. Giovanni Marco Dall'Olio studied biotechnology and bioinformatics in Bolognia, Italy. His PhD was focused on human population genetics, in the Evolutionary Biology group at the Pompeu Fabra University in Barcelona and his primary research goal was the development of methods to characterize events of genetic selection in the human genome. After his PhD, he became research associate at a Cancer Evolutionary Genomics‘s group at the King’s College of London, UK. There, he was interested in the system biology of cancer and in identifying new potential drug targets for this disease.
In 2016 he joined GSK, where he became a core member of a team that intents to dive into the 'data lake' and extract best business values. He explors technology, prototypes new data analysis workflows and data models, interacts with other data scientists, and designs new visualizations. Besides that, he performs target identification in oncology, manages these data and explores and supports new technological solutions for early drug discovery.
Alongside his career in academia and industry, he was (and still is) an author of a blog on bioinformatics and best programming practices (bioinfoblog.it) and a very active member of BioStars, a questions&answers community for bioinformaticians.
3rd Berlin Summer School - Meet Prof. Dr. Dr. Steve Hoffmann
Posted on March 19, 2019

Meet our invited speaker Prof. Dr. Dr. Steve Hoffmann
Dr. Dr. Hoffmann is leader of the 'computational biology of aging' group at the Leibniz Institute on Aging and professor for 'computational biology' at the Friedrich Schiller University Jena.
We are very happy to have Prof. Dr. Dr. Hoffmann at our Summer School. His outstanding career is an inspiration for a lot of young researchers from different fields and he shows how helpful a background in medicine can be in bioinformatics.
Brief Resume
While studying medicine at the Philipps-University Marburg, he finished in 2004 his first diploma in computer science. In 2005 he passed the state examination in medicine and directly started a masters course in bioinformatics at University Hamburg. Together with his M. Sc. in bioinformatics, he received his medical doctor in 2007 from the Heinrich-Heine-University Düsseldorf.
After moving to Leipzig and starting a group leader position at the Leipzig Research Center for Civilization Diseases, he also started working on his second doctorate in bioinformatics, which he finished in 2014. In 2017, he was appointed professor at the Friedrich Schiller University Jena and started his 'computational biology of aging' group at the Leibniz Institute on Aging.
In a bit more than one decade, he received two doctoral degrees, became a professor and headed two research groups. His over 60 publications in journals like Nature, Genome Research, PLoS, NAR, etc. had (and have) a strong impact in the research community and are regularly cited.

3rd Berlin Summer School - Meet Dr. Knut Finstermeier
Posted on March 04, 2019

Meet our invited speaker Dr. Knut Finstermeier
Dr. Finstermeier is Head of Bioinformatics at Emmanuelle Charpentier's Max Planck Unit for the Science of Pathogens.
We are very happy to have Dr. Finstermeier at our Summer School. He nicely shows that a biologist by training can move to the bioinformatics field and even become Head of Bioinformatics of one of the most prestigious research groups these days.
Brief Resume
After he did his masters in biology at Leipzig University, he got his PhD at the Max Planck Institute for Evolutionary Anthropolgy, where he studied the mitochondrial diversity in highly-divergent Strepsirrhini species. He performed pooled hybridization capture experiments of ancient DNA and modern samples.
After his PhD, he set up a new NGS laboratory at the Institute of Laboratory Medicine, Clinical Chemistry and Molecular Diagnostics of the Leipzig University Hospital and teached the protocols and experimental techniques to students and technicians. He implemented and supervised NGS projects (targeted resequencing via long-range PCR in human, ChIP-Seq of human transcription factors, preparation of mate pair libraries from human DNA, and bacterial shot-gun sequencing and assembly) and performed downstream data analysis for most of those projects, including the development of custom analysis approaches.
After a 4-year time-out from academia at a Seattle (WA, USA) based biotech company, where he analyzed B- and T-cell receptors in humans and mice, he started his bioinformatics position at Emmanuelle Charpentier's group.
Now, two years later, he is already Head of Bioinformatics.
RNA-Seq Data Analysis Workshop, February 2019
Posted on February 25, 2019
Our workshop “RNA-Seq Data Analysis - Quality Control, Read Mapping, Visualization and Downstream Analyses” in Berlin this month was a full success. Our two trainers gave the 26 participants from 14 different countries (Lithuania, USA, Germany, UK, Mexico, Norway, Ireland, Russia, Italy, Belgium, France, Denmark, Sweden, United Arabic Emirates) an introduction on how to work with linux and how to analyze RNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
NGS Data Analysis Workshop, January 2019
Posted on January 31, 2019
Our workshop “Next-Generation Sequencing Data Analysis: A Practical Introduction” in Munich this week was a full success. Our two trainers gave the 20 participants from 9 different countries (Germany, Czech Republic, Israel, France, Brazil, UK, Norway, USA, Italy) an introduction on how to work with linux and how to analyze RNA-Seq and DNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Round Robin Trial: Genetic Testing Platform 'Seamless-NGS' obtained full score
Posted on January 23, 2019
"The BRCA1/2 analysis workflow in the Seamless NGS software convinced us with its ease of use and the fast evaluation of the detected variants. We participated successfully in a QuIP round-robin test, achieving a full score (20/20 points), thus confirming the high accuracy of the genetic testing software. Therefore, Seamless NGS is a qualified data analysis tool. With its help, we could successfully establish NGS-based BRCA1/2 testing in our routine diagnostics."
Dr. Tina Unger (Institute of Pathology, Leipzig University Hospital)
Visit our website for the Seamless NGS Software Platform or download the brochure.
Merry Christmas and a Happy New Year
Posted on December 24, 2018
When we here at ecSeq Bioinformatics think of all the benefits of being in our business, we quickly think of our relationships with great customers like you.
Thank you for giving us the chance to do what we enjoy.

Epigenetics Bio-Hackathon at ecSeq
Posted on December 11, 2018
Right now, seven Early Stage Researchers (ESRs) from the Marie Skłodowska-Curie Innovative Training Network EpiDiverse, majoring in biology, are trained by our ESR Adam in using the earlier developed WGBS and DMR calling pipelines. With three ESRs hosted by Prof. Dr. Peter F. Stadler at the bioinformatics chair of Leipzig university and four ESRs at ecSeq Bioinformatics, they are located in walking distance from each other. Thus, all of them have the unique possibility to get trained by experts from academia and industry. Two different views, two different working methods, one goal.
They already analyzed the first 24 bisulfite-treated DNA-Seq samples from pennycress (thlaspi). With 20GB per sample, our high-performances computer clusters ('epi' and 'diverse') were under heavy load. It took 20 hours to map the 24 samples to the thlaspi genome using the tool erne-bs5, while the other tool, segemehl, needed around one week. Calling differentially methylated regions (DMRs) with the tool metilene took ~4h.
Now the ESRs are trained by experts within the EpiDiverse network in how to interprete the results. By overlapping the DMRs with annotations, important meta information such as their genomic position (intron, exon, promoter) is added to the significant regions. Filters, clustering methods and elaborate visualizations will help the researchers to carve out relevant regions that are differentially methylated in stress situations and/or between different environmental conditions.
Meet the PhD students








Some impressions



Annual Workshop Review 2018
Posted on December 11, 2018
We say THANK YOU
The NGS introduction workshop for beginners last week was the last for this year. Thus, we want to thank all participants for their attendance, their thirst for knowledge and their great questions. It was a pleasure for us to give you first impressions of NGS, some insights into the main issues when working with this sequencing method and a starting point for your own analyses.
In our eight public workshops this year we had more than 200 participants from 37 different countries.
The participants of this years workshops came from Austria, Belgium, Chile, Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece, Hong Kong, Israel, Italy, Japan, Jordan, Lebanon, Lithuania, Mexico, Netherlands, New Zealand, Nigeria, Oman, Pakistan, Poland, Qatar, Romania, Russia, Serbia, Singapore, Slovakia, South Africa, Spain, Sweden, Switzerland, Uruguay, United Kigdom and USA.
What courses are planned for 2019?
For 2019 we will have nine public workshops in Leipzig, Berlin, Munich and Freising. If you like to join, please check our upcoming courses in 2019.
Some impressions from 2018





















NGS Data Analysis Workshop, Dec 2018
Posted on December 10, 2018
Our workshop “Next-Generation Sequencing Data Analysis: A Practical Introduction” in Berlin last week was a great event. Our two trainers gave the 20 participants from 11 different countries (Nigeria, Germany, Pakistan, Puerto Rico, Bulgaria, Lebanon, Belgium, Poland, Norway, Czech Republic, Switzerland) an introduction on how to work with linux and how to analyze RNA-Seq and DNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
DNA Methylation Data Analysis, Nov 2018
Posted on December 05, 2018
Our workshop “DNA Methylation Data Analysis” in Berlin last week was a full success. Our two trainers gave the 18 participants from 13 different countries (Germany, Chile, USA, Austria, Czech Republic, Qatar, Russia, France, Belgium, Lebanon, Denmark, Netherlands, Spain) an introduction on how to work with linux and how to analyze Bisulfite-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
NGS Workshop Schedule for 2019
Posted on December 03, 2018
Finally, our workshop schedule for 2019 is online. Based on the availibility of our trainers and extensive feedback from former participants and interested persons, we put together the following courses and dates.
NOTE: Since the Berlin Summer Schools fill up pretty fast every year, you should apply fast to catch your seat.
EpiDiverse Summer School 2018
Posted on November 21, 2018
Bioinformatics for genome and epigenome analysis
During the EpiDiverse Summer School, the Early Stage Researchers (ESRs) of the Innovative Training Network (ITN) 'EpiDiverse' got an introduction to the linux operating system and the method of Bisulfite-Seq for DNA methylation calling. The EpiDiverse pipelines for WGBS data analysis and DMR calling and the internal data management plan were explained in detail. Both were developed earlier by the bioinformatics students at ecSeq in Leipzig. Furthermore, the students were trained in the usage of the pipelines and got an intensive hands-on course in python programming.
Now the ESRs are all set to start analysing the data that will start coming in soon.
EpiDiverse Mid-Term Meeting
Posted on November 20, 2018

ecSeq and EpiDiverse consortium welcome EU project officer in Leipzig to review progress of the ITN project
On Tuesday 13th November 2018, the EpiDiverse consortium came together for the mid-term meeting at Leipzig University. Representatives from all beneficiaries, including the 15 Early Stage Researchers (ESRs), welcomed the responsible project officer Patricia Rischitor from the European Commission’s Research Executive Agency (EC-REA) and spent a complete day to discuss the progress of the EpiDiverse project. After the meeting, the project management team and the project officer were invited to visit ecSeq's offices in the city centre of Leipzig.
As a beneficiary of EpiDiverse, ecSeq supervises one ESR, provides NGS trainings to all ESRs and operates the EpiDiverse high-performance computer clusters.
RNA-Seq Data Analysis Workshop, October 2018
Posted on October 30, 2018
Our workshop “RNA-Seq Data Analysis Workshop: Quality Control, Read Mapping, Visualization and Downstream Analyses” in Berlin last week was a full success. Our three trainers gave the 24 participants from 10 different countries (France, Germany, UK, Israel, Switzerland, Denmark, Italy, New Zealand, Japan, Lithuania) an introduction on how to work with linux and how to analyze RNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Meet ecSeq at the 9th Gene Quantification Event 2019
Posted on October 30, 2018
 Picture talen by Renardo la vulpo - , CC BY-SA 4.0, Link
Picture talen by Renardo la vulpo - , CC BY-SA 4.0, Link
ecSeq will participate the '9th Gene Quantification Event - qPCR dPCR & NGS 2019', which will take place at the Technical University of Munich in Freising from March 18th to 22nd. After the conference we will hold a hands-on workshop to demonstrate how a sophisticated variant analysis can be performed using our genetic testing platform 'Seamless NGS'.
If you are planning on attending this conference as well, feel free to contact us beforehand to arrange a short meeting. We will be happy to present you the newest features of our genetic testing platform and talk about how Seamless NGS can improve your lab routine.
Visit the website of the conference for a detailed agenda of the workshop and further information.
NGS Data Analysis Workshop, October 2018
Posted on October 11, 2018
Our workshop “Next-Generation Sequencing Data Analysis: A Practical Introduction” in Berlin this week was a great event. Our two trainers gave the 19 participants from 9 different countries (Belgium, Germany, Netherlands, Italy, Poland, China, Sweden, UK, Switzerland) an introduction on how to work with linux and how to analyze RNA-Seq and DNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
How-To: Organize Bioinformatics Training Courses
Posted on October 05, 2018

Research in biology has been progressively influenced by big data such as massive genome and transcriptome sequencing data. The handling and analysis of such data require computational skills that usually exceed the abilities of most traditionally trained biologists. In this interesting publication, ecSeq and others discuss the advantages, challenges and considerations for organizing and running bioinformatics training courses of 2–3 weeks in length to introduce biologists to the computational analysis of NGS data.
Reference:
Faria R, Triant D, Perdomo-Sabogal A, Overduin B, Bleidorn C, Bermudez Santana CI, LangenbergerD, Dall’Olio GM, Indrischek H, Aerts J, Engelhardt J, Engelken J, Liebal K, Fasold M, Robb S, Grath S, Kolora SRR, Carvalho T, Salzburger W, Jovanovic V and Nowick K: 'Introducing evolutionary biologists to the analysis of big data: guidelines to organize extended bioinformatics training courses'. Evolution: Education and Outreach (2018)

NGS Data Analysis Workshop, August 2018
Posted on August 27, 2018
Our workshop “Next-Generation Sequencing Data Analysis: A Practical Introduction” in Munich this week was a great event. Our two trainers gave the 20 participants from 10 different countries (Belgium, China, Germany, Qatar, Netherlands, Italy, Austria, Russia, Switzerland, Iran) an introduction on how to work with linux and how to analyze RNA-Seq and DNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Outstanding research - Comprehensive RNA-seq Analysis of Melanoma Samples
Posted on August 17, 2018

Melanoma is one of the solid tumors with the highest mutational burden among solid cancers. A better understanding of how gene expression influences the processes of mutational events in early melanomagenesis harbors potential relevance for diagnostics and therapy. Therefore, researchers around Prof. Dr. med. Manfred Kunz and Dr. Gero Doose performed a comprehensive RNA-seq analysis of laser-microdissected melanocytic nevi and primary melanoma samples. Their findings were published in the renowned journal “Oncogene”.
Reference:
Kunz M, Löffler-Wirth H, Dannemann M, Willscher E, Doose G, Kelso J, Kottek T, Nickel B, Hopp L, Landsberg J, Hoffmann S, Tüting T, Zigrino P, Mauch C, Utikal J, Ziemer M, Schulze HJ, Hölzel M, Roesch A, Kneitz S, Meierjohann S, Bosserhoff A, Binder H, Schartl M: 'RNA-seq analysis identifies different transcriptomic types and developmental trajectories of primary melanomas'. Oncogene (2018)
Dr. Gero Doose is head of research and development at ecSeq Bioinformatics GmbH. He developed RNA folding and protein solubility prediction algorithms before he started working with high-throughput sequencing data. Gero implemented detection pipelines for circularized and trans-spliced transcripts. As an expert for split-read alignment analysis, he also developed a sophisticated method to detect differential alternative splicing.
Round Robin Trial 2017: Genetic Testing Platform 'Seamless-NGS' obtained full score
Posted on August 02, 2018
"The BRCA1/2 analysis workflow in the Seamless NGS software convinced us with its ease of use and the fast evaluation of the detected variants. In 2017 we participated successfully in a QuIP round-robin test, achieving a full score (20/20 points), thus confirming the high accuracy of the genetic testing software. Therefore, Seamless NGS is a qualified data analysis tool. With its help, we could successfully establish NGS-based BRCA1/2 testing in our routine diagnostics."
Dr. Tina Unger (Institute of Pathology, Leipzig University Hospital)
Visit our new website for the Seamless NGS Software Platform or download the brochure.

Epigenetics Hackathon at ecSeq
Posted on August 01, 2018
Right now, three dedicated PhD students, majoring in bioinformatics, are developing a comprehensive software pipeline for the analysis of Bisulfite-Seq data. This software will be used to analyze all incoming bisulfite-treated sequencing data from the EpiDiverse Training Network.
The software will automatically perform the following steps:
- Raw sequence quality control
- Adapter clipping
- Read mapping
- Mapping quality control
- Methylation calling
- DMP calling
- DMR calling
Meet the PhD students


Some impressions


2nd Berlin Summer School, June 2018
Posted on July 03, 2018
Our 2nd Berlin Summer School in NGS Data Analysis in Berlin last week was a full success. Our four trainers gave the 30 participants from 18 different countries (Germany, Singapore, Estonia, Greece, USA, Denmark, Oman, Finland, UK, Uruguay, South Africa, Qatar, Ireland, Slovenia, Netherlands, Lebanon, Italy and Iran) an introduction on how to work with linux and how to analyze DNA-Seq/RNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
New: ecSeq's Genetic Testing Platform
Posted on May 25, 2018
Today we introduce to you our new Genetic Testin Software called Seamless NGS. It is a software platform for automated analysis and management of next-generation sequencing experiments. By providing easy access to advanced bioinformatics protocols, it shortens the time from data generation to interpretation.
Visit our new website for the Seamless NGS Software Plafrom.
Additional NGS Introduction Course in October 2018
Posted on April 27, 2018
Due to an extraordinary run on our NGS Introduction workshops this year and astounding long waiting lists, we decided to offer an additional NGS Introduction course in October in Berlin.

NGS Data Analysis Workshop, April 2018
Posted on April 27, 2018
Our workshop “Next-Generation Sequencing Data Analysis: A Practical Introduction” in Munich this week was a full success. Our two trainers gave the 20 participants from 11 different countries (Sweden, Belgium, Spain, Slovakia, Russia, Austria, UK, France, Netherlands, Germany, Serbia) an introduction on how to work with linux and how to analyze RNA-Seq and DNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
RNA-Seq Data Analysis Workshop, March 2018
Posted on March 21, 2018
Our workshop “RNA-Seq Data Analysis Workshop: Quality Control, Read Mapping, Visualization and Downstream Analyses” in Berlin last week was a full success. Our three trainers gave the 30 participants from 10 different countries (Germany, USA, Poland, Czech Republic, United Kingdom, Romania, France, Lithuania, Mexico and Switzerland) an introduction on how to work with linux and how to analyze RNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Merry Christmas and a Happy New Year
Posted on December 22, 2017
During the holiday season, our thoughts turn gratefully to those who have made our success possible.
It is in this spirit that we say thank you and best wishes for the holidays and New Year.

NGS Data Analysis Workshop, December 2017
Posted on December 12, 2017

Our workshop Next-Generation Sequencing Data Analysis: A Practical Introduction in Munich last week was a full success. Our three trainers gave the 20 participants from 11 different countries (Austria, Belgium, Denmark, France, Germany, Malta, Netherlands, Sweden, Taiwan, Thailand and USA) an introduction on how to work with linux and how to analyze NGS data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.

DNA Methylation Data Analysis, 2017
Posted on December 07, 2017

Our workshop “DNA Methylation Data Analysis” in Berlin last week was a full success. Our two trainers gave the 25 participants from 12 different countries (Belgium, Denmark, France, Germany, Israel, Japan, New Zealand, Norway, Saudi Arabia, Spain, Sweden and Switzerland) an introduction on how to work with linux and how to analyze Bisulfite-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Open PhD Position
Posted on October 18, 2017
Inferring genomic information from bisulfite sequencing data
- PhD Supervisor(s): Dr. David Langenberger, Prof. Dr. Peter F. Stadler
- Host Institution: ecSeq Bioinformatics GmbH
in collaboration with the Chair for Bioinformatics University Leipzig - Duration: 36 months
- Fixed start date: 1 April 2018
- Planned secondment(s): Gregor Mendel Institute of Molecular Plant Biology (Vienna, Austria), IGA Technology Services SRL (Udine, Italy) and Roche Diagnostics GmbH (Mannheim, Germany)
Background
DNA methylation variants can arise spontaneously, they can be under genetic control or they can be induced by the environment. In plants, some DNA methylation variants are stable across many generations whereas other variants are very transient. A good understanding of the transgenerational dynamics of DNA methylation variants is essential to understand their impact on heritable traits and their effect on adaptation. Current insight in the transgenerational dynamics of DNA methylation is limited to very few model plant species, but it is predicted that these dynamics are not constant among different plants. For instance, plant reproduction mode can have a large effect because asexual reproduction bypasses some of the epigenetic resetting mechanisms that normally occur during sexual reproduction. Adaptive differences in transgenerational stability may also differ between species with different life spans or from habitats of different environmental predictability.
Your PhD Project
In this project, we aim to investigate differences in DNA methylation dynamics between species with different life history traits. In close collaboration with other bioinformaticians and biologists, a best-practice pipeline for the analysis of plant bisulfite data will be developed, benchmarked, and provided to be applied to the Next-Generation Sequencing (NGS) data generated in the frame of different EpiDiverse projects. In addition, novel algorithms will be devised and implemented to better detect DNA methylation variants and search for important methylation haplotypes to better understand the epigenetic regulation. The special constellation of working at a bioinformatics company in very strong cooperation with the chair of bioinformatics at the university offers the unique possibility of gaining benefits from both: The clearly structured style of the work in a company will help implementing novel ideas from academia in the light of recent publications, resulting in a strong focus and high productivity.
Your Qualifications
We seek a bright, highly motivated, and enthusiastic bioinformatician, or computer scientist with significant interest in solving biologically motivated questions. Excellent programming skills with knowledge of at least one of the following programming languages are mandatory: Python, R, or C/C++/C#. A strong background in Next-Generation Sequencing (NGS) and/or (epi)genetics data analyses are an advantage. A high standard of spoken and written English is required, as are good quantitative and analytical capabilities, excellent interpersonal and communication skills, and the ability to work independently as well as part of a team.
Eligibility criteria
A university degree in a relevant field is required. Eligibility criteria of EU H2020 ITN programmes apply: candidates must be, at the time of recruitment by the host organisation, in the first four years (full-time equivalent) of their research careers and have not yet been awarded a doctoral degree. This is measured from the date when they obtained the degree which would formally entitle them to embark on a doctorate. Eligible candidates may be of any nationality but must not, at the time of recruitment, have resided or carried out their main activity (work, studies, etc) in the country of their host organisation for more than 12 months in the 3 last years immediately prior to the reference date.
How to apply
You may apply by providing a letter of motivation, a detailed CV and contact information of up to three references, all as a single pdf file!
Send the file by email:
- to: info@epidiverse.eu
- Cc: support@ecseq.com
- Subject: EpiDiverse-Application RP-3
If you have questions, you can contact support@ecseq.com.
Keydates
- Application deadline:
24 November 2017 - Interviews: December 2017
- Starting date: 1 April 2018
About EpiDiverse
EpiDiverse - Epigenetic Diversity in Ecology - is a Marie Skłodowska-Curie Innovative Training Network aimed at the study of epigenetic variation in wild plant species. This network joins academic groups from ecology, molecular (epi)genetics and bioinformatics with life science companies to explore epigenetic mechanisms and their adaptive relevance in natural plant populations.

The cross-disciplinary research program investigates how epigenetic mechanisms, in interaction with environments and transposable elements, contribute to variation that is relevant for the adaptive capacity of plants. By applying epigenomic research tools to a selection of different wild plants (including long-lived trees, annual and asexually reproducing species) we investigate how epigenetic mechanisms as well as their ecological relevance differs between plant species. Understanding the epigenetic contribution to adaptive capacity will help to predict species responses to global warming and can open new directions for sustainable agriculture and crop breeding.
The EpiDiverse consortium will train 15 Early Stage Researchers to become expert eco-epigeneticists, equipping them with the interdisciplinary skills - molecular, (epi)genomic, ecological and bioinformatics - to successfully tackle this new research area. EpiDiverse training will emphasize empirical and informatics skills to become fluent and creative in extracting knowledge from big ‘omics data in natural contexts.
EpiDiverse is funded by the EU Horizon 2020 programme and involves 12 partners from academia, non-profit organizations and industry located in the Netherlands, Germany, France, Spain, Czech Republic, Italy and Austria.
For further Information check the official website of EpiDiverse.
RNA-Seq Data Analysis Workshop, October 2017
Posted on October 15, 2017

Our workshop “RNA-Seq Data Analysis Workshop: Quality Control, Read Mapping, Visualization and Downstream Analyses” in Leipzig last week was a full success. Our three trainers gave the 20 participants from 9 different countries (Norway, Germany, China, USA, UK, Czech Republic, Belgium, Netherlands, Qatar) an introduction on how to work with linux and how to analyze RNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
1st Berlin Summer School, September 2017
Posted on September 16, 2017

Our 1st Berlin Summer School in NGS Data Analysis in Berlin last week was a full success. Our four trainers gave the 30 participants from 16 different countries (China, Germany, Poland, Austria, France, Belgium, Czech Republic, Denmark, United Kingdom, Norway, Switzerland, Slovenia, Netherlands, Japan, Réunion and Sweden) an introduction on how to work with linux and how to analyze DNA-Seq/RNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.



ecSeq Bioinformatics Joins Forces with Seamless NGS
Posted on August 30, 2017

You may have already heard of ecSeq Bioinformatics' cooperation with Seamless NGS, a biomedical software manufacturer and developer of tailored NGS solutions. We are pleased to inform you that last month there has been a merger between the two companies.
Seamless NGS has developed a unique portfolio of cutting-edge NGS software solutions which are already used in diagnostic laboratories and research facilities. They specialised on high-performance analyses of DNA, RNA and epigenetic markers for different diseases such as cancer.
We have joined forces with Seamless NGS to offer you a more complete portfolio for your NGS profiling. It is our mission to provide you with the best bioinformatics technologies and we believe we can create significant benefits for you in terms of convenience, cost-efficiency, and quality of results.
We are dedicated to maintaining and increasing the quality of innovation, support and service you expect from ecSeq Bioinformatics and Seamless NGS. We look forward to providing you with the most complete and advanced bioinformatics solutions for your NGS experiment.
Bringing the power of Big Data to biomedical researchers
Posted on July 28, 2017
Great article about ecSeq Bioinformatics's work and vision in the current issue of 'Healthcare Journal of Baton Rouge'. Thanks to Dr. Claudia S. Copeland (LinkedIn Profile) for this understandable article about a very complex topic
Read Flipping Book Version
Read PDF Version
NGS Data Analysis Workshop, June 2017
Posted on June 30, 2017

Our workshop “Next-Generation Sequencing Data Analysis: A Practical Introduction” in Frankfurt this week was a full success. Our two trainers gave the 18 participants from 7 different countries (Germany, Croatia, South Korea, Netherlands, Israel, Czech Republik and Finland) an introduction on how to work with linux and how to handle RNA-Seq and DNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Additional RNA-Seq Course 2017
Posted on June 29, 2017
Additional RNA-Seq Course in October 2017
Due to an extraordinary run on our "1st Berlin Summer School" in September in Berlin and an astounding long waiting list, we decided to offer an additional RNA-Seq course with almost the same topics in October in Leipzig.

NGS Data Analysis Workshop, May 2017
Posted on May 11, 2017

Our workshop “Next-Generation Sequencing Data Analysis: A Practical Introduction” in Munich last week was a full success. Our two trainers gave the 20 participants from 8 different countries (Germany, USA, Saudi Arabia, UK, Romania, Belgium, Slovenia and Turkey) an introduction on how to work with linux and how to analyze RNA-Seq and DNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Five Year Anniversary
Posted on April 15, 2017
CELEBRATING OUR 5TH YEAR ANNIVERSARY TODAY!
Today marks 5 years exactly that ecSeq Bioinformatics entered the highly competitive and innovative world of life sciences. In the last 5 years we have learned a lot, shared a lot, taught a lot and built a lot. We have seen substantial growth take place with the sequencing technology. We truly believe this technology and industry will develop rapidly in the coming years.
We have had the pleasure of working with some amazing clients, building great solutions and relationships that extend well beyond typical client/contractor engagements. We also want to send a special shout out to our sequencing partners who give us a much needed helping hand to deliver high quality solutions for our clients.
We have some exciting things in the works for the remainder of the year, so stay tuned. As we continue our journey, we are excited to see what the next 5 years and beyond will bring.
RNA-Seq Data Analysis Workshop, March 2017
Posted on March 25, 2017

Our workshop “RNA-Seq Data Analysis Workshop: Quality Control, Read Mapping, Visualization and Downstream Analyses” in Berlin last week was a full success. Our three trainers gave the 25 participants from 11 different countries (Germany, USA, Qatar, Netherlands, Hungary, Belgium, China, United Kingdom, Poland, Croatia and Italy) an introduction on how to work with linux and how to analyze RNA-Seq data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an equal level.
Joint project with Fraunhofer IME
Posted on February 08, 2017

Rubber from dandelions
In recent years the Russian dandelion Taraxacum koksaghyz, a previously neglected wild plant, has been investigated to be re-introduced as a valuable rubber crop that grows in moderate climates, like in Germany. The milky sap of the dandelion shows similarities to that of the rubber tree, and most importantly contains caoutchouc, the raw material for rubber production.
Together with the Fraunhofer Institute for Molecular Biology and Applied Ecology IME, ESKUSA GmbH and hortilab, ecSeq GmbH will generate a high-resolution genetic map of Russian dandelion.

Dr. Christian Schulze Gronover (Fraunhofer IME) received the 2015 Joseph von Fraunhofer Prize for his work on the Russian dandelion and its application as a source of natural rubber. He is the coordinator of the project, which is funded by the German Federal Ministry of Education and Research (BMBF).

Dr. Fred Eickmeyer (ESKUSA GmbH) specialized on breeding of dandelion. He wants to optimize and establish Russian dandelion to an alternative raw material for natural rubber.

Cooperation Agreement with Emmanuelle Charpentier's Laboratory
Posted on February 01, 2017
Today we signed a cooperation agreement with Prof. Dr. Emmanuelle Charpentier.
We are looking forward to this great partnership.
Prof. Dr. Emmanuelle Charpentier is best known for her role in deciphering the molecular mechanisms of the bacterial CRISPR-Cas9 immune system and repurposing it into a tool for genome editing. In particular she uncovered the mechanism beyond the maturation of non-coding RNA key for the CRISPR-Cas9 function. In collaboration with Jennifer Doudna's laboratory, Charpentier's laboratory showed that Cas9 could be used to make cuts in any DNA sequence desired. The method they developed involved the combination of Cas9 with easily created synthetic "guide RNA" molecules. Researchers worldwide have employed this method successfully to edit the DNA sequences of plants, animals, and laboratory cell lines.
Sources: Picture and text from wikipedia.org; Video from youtube.com (DFG channel)
Outstanding research - New macromolecule with influence on Burkitt lymphoma detected
Posted on January 27, 2017
Dr. Gero Doose is head of research and development at ecSeq GmbH. He developed RNA folding and protein solubility prediction algorithms before he started working with high-throughput sequencing data. Gero implemented detection pipelines for circularized and trans-spliced transcripts. As an expert for split-read alignment analysis, he also developed a sophisticated method to detect differential alternative splicing.

The transcription factor MYC plays a key role in carcinogenesis. Together with scientists around Dr. Ingram Iaccarino (Christian Albrechts University) and Dr. Dr. Steve Hoffmann (University of Leipzig), Dr. Doose discovered a long non-coding RNA that is able to modulate MYC’s transcriptional network. This finding therefore has the potential to open new therapeutic opportunities for MYC associated cancers. The results were published in the renowned journal “Proceeding of the National Academy of Science of the United States of America” (PNAS).
Reference:
Doose G, Haake A, Bernhart SH, López C, Duggimpudi S, Wojciech F, Bergmann AK, Borkhardt A, Burkhardt B, Claviez A, Dimitrova L, Haas S, Hoell JI, Hummel M, Karsch D, Klapper W, Kleo K, Kretzmer H, Kreuz M, Küppers R, Lawerenz C, Lenze D, Loeffler M, Mantovani-Löffler L, Möller P, Ott G, Richter J, Rohde M, Rosenstiel P, Rosenwald A, Schilhabel M, Schneider M, Scholz I, Stilgenbauer S, Stunnenberg HG, Szczepanowski M, Trümper L, Weniger MA; ICGC MMML-Seq Consortium, Hoffmann S, Siebert R, Iaccarino I: 'MINCR is a MYC-induced lncRNA able to modulate MYCs transcriptional network in Burkitt lymphoma cells'. PNAS, 112(38):E5261-70. (2015)
Next-Generation Sequencing Data Analysis: A Practical Introduction, December 2016
Posted on December 13, 2016

Our workshop “Next-Generation Sequencing Data Analysis: A Practical Introduction” in Munich last week was a full success. Our two trainers gave the twenty participants from eight different countries an introduction on how to work with linux and how to analyze NGS data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
Keynote at bionection 2016 in Halle (Saale)
Posted on September 23, 2016

bionection is an international event for technology transfer in life sciences. Thus, participants from industry and academia will present and discuss their latest inventions in this hot research field. The venerable Leopoldina provides an amazing scene for inspiring discussions, extraordinary thoughts and exchange of innovative ideas.
Besides Thermo Fischer and Thyssenkrupp Industrial Solutions, ecSeq’s CEO Dr. David Langenberger will be one of the three keynote speakers at the event. On the second day of the conference, David will talk about today's challenges in NGS bioinformatics. Our business area, NGS bioinformatics, fits very well into the overall aim of bionection, where the latest progress in medical and bio-technologies, as well as current questions in life sciences will be presented.
The German National Academy of Sciences was founded in 1652. Since then it is “dedicated to the advancement of science for the benefit of humankind and to the goal of shaping a better future”. ecSeq GmbH is honored to speak in such history-charged halls in Halle (Saale).
Nature Genetics - DNA Methylome Analysis
Posted on September 15, 2016

Great News:
The two trainers of our upcoming epigenomics workshop 'DNA Methylation Data Analysis' got their research work published in Nature Genetics.
Dr. Helene Kretzmer and Dr. Christian Otto identified differentially methylated regions in Burkitt and follicular lymphoma using exactly the bisulfite sequencing data analysis method that they will teach in the workshop.
Publication:
Kretzmer et al.: 'DNA methylome analysis in Burkitt and follicular lymphomas identifies differentially methylated regions linked to somatic mutation and transcriptional control', Nature Genetics (2015)
 |
Dr. Helene Kretzmer (University Leipzig) is working on DNA methylation analyses using NGS since 2011. She is responsible for the bioinformatics analysis of the MMML-Seq study of the International Cancer Genome Consortium (ICGC). | |
 |
Dr. Christian Otto (CCR Bio-IT) is one of the developers of bisulfite read mapping tool segemehl and is an expert on implementing efficient algorithms for NGS data analysis. |
ecSeq's NGS Experts
Posted on July 28, 2016

Up-to-date expert knowledge is the key to generating impactful biological insights.
To address this unavoidable need, we established an extensive network of outstanding scientists, covering all research fields relevant to NGS. Together with a project-specific expert panel ('think tank'), we will analyze your data in a scientifically proven manner to provide you with meaningful, knowledge-driven results.
-
Our expert-network consists of 46 scientists:
13 Professors
31 PhDs
3 PhD students
-
Together with our expert network, we can offer the advantages of both, academia and industry:
scientifically sound analyses
approved and interpreted by experts
timely delivery
Go through the complete list of scientists and check the extraordinary experts in our expert-network:
Start a knowledge-driven project with ecSeq Bioinformatics
Next-Generation Sequencing Data Analysis: A Practical Introduction, June/July 2016
Posted on July 06, 2016

Our workshop “Next-Generation Sequencing Data Analysis: A Practical Introduction” in Munich two weeks ago was a full success. Our two trainers gave the twenty participants from nine different countries an introduction on how to work with linux and how to analyze NGS data correctly. The gained knowledge will allow the researchers to start their own analyses and it will help them to discuss their bioinformatics problems or questions with bioinformaticians on an almost equal level.
For everyone who was not able to attend we will organize a second workshop in December 2016. It will be at the same location in Munich and it will cover the exact same topics. Perfect for beginners.
Lab journal with a portrait of ecSeq GmbH
Posted on May 04, 2016

We recently received a visit by Tobias Hoffmann, a journalist from the German "Laborjournal" (German for lab journal). In its latest issue (from May 2nd, 2016) ecSeq GmbH and its founders, Dr. David Langenberger and Dr. Mario Fasold, are portrayed in a double-page article.
The article talks about our two founders, how they gained their doctorates from professor Stadler in Leipzig and their way to ecSeq GmbH. One of our business areas are workshops which train the participants how to analyze NGS data. We want to impart a discretionary competence as well as evaluation skills with the courses. Public workshops take place in Munich, Berlin, Prague or Oxford with about one third of the participants having an industrial (non-academic) background. This knowledge is important because nowadays every research application contains NGS analyzes.
Secondly, Hoffmann introduces our network of 45 highly skilled NGS experts to the readers. During project consulting ecSeq GmbH accompanies NGS projects which require complex bioinformatics solutions. Customers can book nearly everything: the analyses, the software or the complete planning of an experiment. All NGS experts are working in the background together with ecSeq GmbH.
Of course, the author asks the follow up question: 'Why should customers approach ecSeq and not the experts directly?' The answer is pretty simple: 'Because academic researchers with their reservations are often not very open-minded about inquiries from companies.' With ecSeq GmbH as intermediary, a trustful and well-managed collaboration is possible. This separation of industry and academia is very important for both, the companies and the researchers in our network.
We thank the "Laborjournal" for this great introduction of our company! You can get your free copy of the "Laborjournal" at universities and research facilities or on their website.
Let's Talk About Starting Your Own Company - ecSeq at Life Sciences Career Day Dresden
Posted on April 26, 2016

At the Life Sciences Career Day Dresden students from biology, biotechnology, biochemistry, medicine & pharmacy can get an impression about how their future could look like. We at ecSeq GmbH are very honored to represent the career path of a start-up company in the field of life sciences and to embold young scientists to realize their own ideas!
Deciding where to go after university is by far not trivial and has to be well thought out. Researchers have to realize that an academic career with the aim to get a professorship or the industrial path as an employee are not the only possibilities. We will explain and picture an interesting, but challenging middle course, the entrepreneurship. During our presentation we will tell the exciting story about starting our own company. We will describe how to turn the dream of a business into reality, talk about the initial period and finally, demonstrate how our daily work routine looks like. Afterwards, all upcoming questions will be answered.
The Life Science Career Day takes place on June 3rd, 2016 at the "Hörsaalzentrum" in Dresden, Germany.
It is organized by the biotechnology students initiative btS. We are looking forward to meet enthusiastic students who would like to discuss bioinformatics and its future with us.
These questions will be answered:
- What is Next-Generation Sequencing?
- What role does it already play in research and the daily clinical business?
- Can a start-up in the field of NGS bioinformatics be successful?
Membership in life science cluster 'biosaxony'
Posted on April 01, 2016
It is always important to have a good network of partner corporations and institutes to learn from each other. To this end, ecSeq GmbH is now a member of biosaxony, the cluster of the biotechnology and life sciences sector in Saxony. The organization hosts projects with participating scientists and companies, provides its members useful services and know-how, has the aim to develop the branch and strengthens the business location Saxony. It combines members from areas of economy, science and politics like young start ups, universities, research institutes, municipalities as well as large-scale enterprises.
We are looking forward to an interesting exchange with the Saxon biotechnology network!
RNA Expert - Dr. Martin Smith
Posted on March 21, 2016

We are very pleased to introduce Dr. Martin Smith as a member of our NGS Expert Network.
Dr. Martin Smith is a computational biologist with a strong background in biology. As a child he grew up in Canada and he always dreamt of becoming a biologist. He majored as an undergraduate in microbiology and immunology at the University of Montreal, but he quickly realised this wasn’t his field. Switching to the emerging field of bioinformatics was an enthusiastic yet challenging decision for him, as it required rewiring his brain around abstract informatics concepts and discrete mathematics.
To this day, he is very passionate about bioinformatics and would recommend a cross-disciplinary career path to anyone. He has worked in 5 different cities across 3 different continents, namely Montreal (University of Montreal); Quebec City (Infectious Disease Research Centre—CHUL); Brisbane (Institute for Molecular Bioscience); Leipzig (Interdisciplinary Center for Bioinformatics); and Sydney (Garvan). He received his doctorate in Genomics and Computational Biology from the University of Queensland in 2012. His doctor's thesis was supervised by Prof. Dr. John Mattick and Prof. Dr. Peter F. Stadler.
His research revolves around characterising the “regulome”, i.e. the biological mechanisms that control how genes are activated and repressed. He is particularly interested in how non-protein coding regions of the genome can regulate gene expression at the level of RNA.
Evaluation: Annual Survey - Public NGS Workshops
Posted on February 29, 2016

Around the turn of the year (2015/2016), we asked the question:
What courses would you like us to prepare for 2016?
We analyzed all votes (~200 single votes) and below you can find the result of the survey. Based on your voting, we put together the workshop agenda for 2016.
We thank all attendees! We hope that our agenda fits your expectations!
Assembly Expert - Dr. Alexander Donath
Posted on February 25, 2016

We are very pleased to introduce Dr. Alexander Donath as a member of our NGS Expert Network.
Dr. Alexander Donath is head of section for Computational Genomics at the Zoological Research Museum Alexander Koenig. Since 2011 he is working on NGS data analysis and de novo assembly of non-model organisms. He is involved in a number of large-scale transcriptome and genome research projects, for example 1KITE and i5k.
Assembly Workshop 2016
Besides being part of the NGS Expert Network, he also designed our hands-on workshop for de novo transcriptome analysis, which will cover assembly and differential gene expression analysis in non-model organisms.
Outstanding Research
In 2014 Dr. Alexander Donath co-authored the Science paper Phylogenomics resolves the timing and pattern of insect evolution, which was honoured as a research highlight by Nature Reviews Genetics.
'... In this research, novel approaches were used to produce a robust phylogenomic tree of insects, which will now serve as a reference point for future research.' (Nature)

|
'... The study authors, who are part of an international consortium working on the 1K Insect Transcriptome Evolution (1KITE) project, began their work in 2011 in hopes of using newly available sequencing technology to clarify insect relationships previously only investigated using morphological evidence or smaller molecular datasets. They sequenced transcriptomes from 103 insect species distributed across all living insect orders. They also drew data from previously published whole-genome sequences of 14 arthropod species, as well as from transcriptomes of 27 additional species. They then narrowed down their genetic data to 1,478 protein-coding genes that are present in all of the species analyzed. ...' from TheScientist |
Reference:
Bernhard Misof, Shanlin Liu, Karen Meusemann, Ralph S. Peters, Alexander Donath, Christoph Mayer, Paul B. Frandsen, Jessica Ware, Tomáš Flouri, Rolf G. Beutel, Oliver Niehuis, Malte Petersen, Fernando Izquierdo-Carrasco, Torsten Wappler, Jes Rust, Andre J. Aberer, Ulrike Aspöck, Horst Aspöck, Daniela Bartel, Alexander Blanke, Simon Berger, Alexander Böhm, Thomas R. Buckley, Brett Calcott, Junqing Chen, Frank Friedrich, Makiko Fukui, Mari Fujita, Carola Greve, Peter Grobe, Shengchang Gu, Ying Huang, Lars S. Jermiin, Akito Y. Kawahara, Lars Krogmann, Martin Kubiak, Robert Lanfear, Harald Letsch, Yiyuan Li, Zhenyu Li, Jiguang Li, Haorong Lu, Ryuichiro Machida, Yuta Mashimo, Pashalia Kapli, Duane D. McKenna, Guanliang Meng, Yasutaka Nakagaki, José Luis Navarrete-Heredia, Michael Ott, Yanxiang Ou, Günther Pass, Lars Podsiadlowski, Hans Pohl, Björn M. von Reumont, Kai Schütte, Kaoru Sekiya, Shota Shimizu, Adam Slipinski, Alexandros Stamatakis, Wenhui Song, Xu Su, Nikolaus U. Szucsich, Meihua Tan, Xuemei Tan, Min Tang, Jingbo Tang, Gerald Timelthaler, Shigekazu Tomizuka, Michelle Trautwein, Xiaoli Tong, Toshiki Uchifune, Manfred G. Walzl, Brian M. Wiegmann, Jeanne Wilbrandt, Benjamin Wipfler, Thomas K. F. Wong, Qiong Wu, Gengxiong Wu, Yinlong Xie, Shenzhou Yang, Qing Yang, David K. Yeates, Kazunori Yoshizawa, Qing Zhang, Rui Zhang, Wenwei Zhang, Yunhui Zhang, Jing Zhao, Chengran Zhou, Lili Zhou, Tanja Ziesmann, Shijie Zou, Yingrui Li, Xun Xu, Yong Zhang, Huanming Yang, Jian Wang, Jun Wang, Karl M. Kjer, Xin Zhou: 'Phylogenomics resolves the timing and pattern of insect evolution', Science 346 (6210): 763-767 (2014).
small RNA-Seq Expert - Prof. Dr. Manja Marz
Posted on January 19, 2016

We are very pleased to introduce Prof. Dr. Manja Marz as a member of our NGS Expert Network.
Prof. Dr. Manja Marz studied biology and computer science at the University of Leipzig, and received her Ph.D. in bioinformatics from the University of Leipzig in 2009. After leading a Group at the Philipss-University of Marburg and Junior-Professorship at the Friedrich-Schiller-University Jena, she got two calls for professorships at University of Lübeck and Friedrich Schiller University Jena in 2014. Since 2015 she holds a full professorship at Friedrich Schiller University Jena. She is also Founding member of MSCJ (Michael Stifel Zentrum Jena for Data-Driven and Simulation Science), Board member of ZAJ (Aging Research Center Jena), Founding and board member of FIFI (Fördervereinverein des Instituts für Informatik) and Group leader at Leibniz Institute for Age Research – Fritz Lipmann Institute.
Her research interest include High Throughput Sequencing Analysis, Comparative Genomics, Identification and Annotation of Non-coding RNAs, Bioinformatic Analysis and System Biology of Viruses, Coevolution of Proteins and RNAs, Algorithmic Bioinformatics and Phylogenetic Analysis.
Happy New Year 2016
Posted on December 30, 2015
With 2015 coming to an end we can't help but feel a little nostalgic and look back at what the past twelve months have brought us. We couldn't be happier with what we do and we have all of you, our loyal customers to thank for our success.
During the past 360 or so days, we have been hard at work to train you in NGS data analysis and analyze your NGS data. We are extremely thankful for all the support and word-of-mouth advertising for our services.
We are all set for 2016 and can't wait to see what the New Year is going to bring to the NGS world.
Wishing you a Happy Holiday and a joyful New Year.
Best wishes from the complete team at ecSeq GmbH.

Annual Survey - Public NGS Workshops
Posted on December 10, 2015

This year, again, we offered several hands-on bioinformatics NGS workshops. We widened our range and went to different venues across Europe in order to make our service available to more and more interested researchers. Besides basic NGS introduction courses, we always try to extend our portfolio to cover further NGS applications.
Our question: What courses would you like us to prepare for 2016?
Fill out our survey today and win a 50% off voucher for one of our NGS workshops!
RNA-Seq Expert - Prof. Dr. Ivo Hofacker
Posted on November 04, 2015

We are very pleased to introduce Prof. Dr. Ivo Hofacker as a member of our NGS Expert Network.
Since 2010 Prof. Dr. Ivo Hofacker holds a double professorship at the faculties of Chemistry and Computer Science. He is currently head of the Research Group Bioinformatics and Computational Biology.
His scientific interests fit mostly under the computational biology umbrella. Much of his work focuses on RNA Bioinformatics, the most visible part perhaps being the Vienna RNA Package for prediction and comparison of RNA secondary structures, which he and his coworkers have developed for over 15 years.

Conntect with us on Facebook
Posted on November 02, 2015

Check out our Facebook page and learn more about our products, our hands-on workshops, our research topics and our expertise in NGS data analysis. Share your ideas and stay conntected.
In our Facebook group 'Next-Generation Sequencing Data Analysis' you will learn about upcoming events, open jobs, and interesting information about the rapidly growing field of Next-Generation Sequencing. So connect with us and discover the world of ecSeq Bioinformatics.

RNA-Seq Expert - Prof. Dr. Rolf Backofen
Posted on October 30, 2015

We are very pleased to introduce Prof. Dr. Rolf Backofen as a member of our NGS Expert Network.
Prof. Dr. Rolf Backofen studied computer science at the University of Erlangen, and received his Ph.D. in computer science from the University of Saarland in December 1994, where he worked at the German Research Center for Artificial Intelligence (DFKI). He received his habilitation from the University Munich (LMU) in February 2000. He was holding the chair for bioinformatics at the University of Jena from November 2001 till June 2005. In 2004 he became the holder of the chair for Bioinformatics at the University of Freiburg, Institute of Computer Science.
His research interest include constraint programming, structure prediction in simplified protein models, investigation of protein energy landscapes, detection of RNA sequence/structure motifs, prediction and evaluation of alternative splice forms, description and detection of regulatory sequences.
Nature Genetics - DNA Methylome Analysis
Posted on October 09, 2015
Great News:
The two trainers of our upcoming epigenomics workshop *DNA Methylation Data Analysis' got their research work published in Nature Genetics.
Helene Kretzmer and Dr. Christian Otto identified differentially methylated regions in Burkitt and follicular lymphoma using exactly the bisulfite sequencing data analysis method that they will teach in the workshop.
Publication:
Kretzmer et al.: 'DNA methylome analysis in Burkitt and follicular lymphomas identifies differentially methylated regions linked to somatic mutation and transcriptional control', Nature Genetics (2015)
|
Helene Kretzmer (University Leipzig) is working on DNA methylation analyses using NGS since 2011. She is responsible for the bioinformatics analysis of the MMML-Seq study of the International Cancer Genome Consortium (ICGC). | |
|
Dr. Christian Otto (CCR Bio-IT) is one of the developers of bisulfite read mapping tool segemehl and is an expert on implementing efficient algorithms for NGS data analysis. |
Introducing Knowledge-Driven NGS Solutions
Posted on September 16, 2015

Nobody can be an expert in everything. However, biomedical projects increasingly demand expert knowledge in more than a single field. In fact, up-to-date expert knowledge is key to generating impactful biological insights.
To this end, we established an extensive network of outstanding scientists, covering various research fields relevant to NGS. The idea is that, together with the customer, we assemble a panel of experts that is able to develop an optimal strategy for the specific challenges of the project (see our white paper).
Our bioinformatics team can then rapidly implement a software solutions since we already have a library of analysis modules for many NGS applications.
Get more information here.
Enabling end-to-end NGS solutions through our partnership with AllGenetics
Posted on April 07, 2015

We are now able to offer end-to-end analysis solutions for the NGS applications RNA-Seq, small-RNA-Seq, and DNA-seq. This includes essential steps from sample preparation and sequencing to extensive bioinformatics data analysis. This is enabled by our partnership with AllGenetics, a biotech company specialized in NGS research projects. Our partnership with AllGenetics ensures that projects can be completed smoothely and effectively from project design to data analysis.
Upcoming NGS Workshop in Prague, CZ
Posted on March 19, 2015

We happily announce that the next NGS workshop will take place September 7-11 in Prague. The course entitled 'PRAGUE SUMMER SCHOOL - NEXT-GEN SEQ DATA ANALYSIS' spans a whole week and is organized by SEQme in cooperation with ecSeq Bioinformatics. The workshop specifically addresses the needs of beginners in the field of NGS bioinformatics.
Please click here to visit the workshop website.
Meet us at the Illumina User Group Meeting, EMBL
Posted on February 26, 2015
The Illumina User Group Meeting (UGM) Central Europa is taking place on March 10-11 at the EMBL in Heidelberg, Germany. Dr. Mario Fasold will present new possibilities for bioinformatic analysis of RNA-seq data and also be available for discussions on the afternoon of March 11. Please contact us if you would like to schedule a meeting there.
Upcoming: NGS Workshop in Leipzig, Germany
Posted on November 27, 2014
In March 2015 our NGS data analysis are again taking place in Leipzig. The workshop has been redesigned and adapted to the needs of beginners in the field of NGS bioinformatics. There are now two optional courses on RNA-seq data analysis (model and non-model organisms). Building on the knowledge covered in the main course allows us to cover the many relvant aspects of downstream RNA-seq analyses.
NGS Workshop at 'Programming for Evolutionary Biology' in Bogota, Colombia
Posted on October 29, 2014
In this intensive course, students will learn how to survive in a Linux environment, get hands-on experience in two widely used programming languages (Perl and R), and statistical data analysis. The classes will be given by experts in the field and consist of lectures and exercises with the computer. The aim of the course is to provide the students with the necessary background and skills to perform computational analyses with a focus on solving research questions related to genomics and evolution. The philosophy of the course will be “learning by doing”, which means that the computational skills will be taught using examples and real data from evolutionary biology for the exercises. During the course, students will also propose projects of their own interest and perform them as final projects in small groups under the supervision of a teaching assistant. This November school the school goes to Colombia. It is open for students from all countries, but specially, from the Americas. It is mainly targeted toward PhD students, MSc students and Postdocs of evolutionary biology or related research fields with no or little programming experience who want to become proficient in computational evolutionary biology in a couple of weeks.
Link to the workshop page.
Upcoming: NGS Workshop in Oxford, UK
Posted on October 27, 2014
The purpose of this workshop is to get a deeper understanding in Next-Generation Sequencing (NGS) with a special focus on bioinformatics issues. Advantages and disadvantages of current sequencing technologies and their implications on data analysis will be discovered. The participants will be trained on understanding their own HTS data, finding potential problems/errors therein and finally performing their own analyses using open source tools. In the course we will use a RNA-seq dataset from the current market leader, Illumina.
Link to the workshop page.
New Publication in Nature Communications
Posted on October 08, 2014

Sarah A. Munro, Steven P. Lund, P. Scott Pine, Hans Binder, Djork-Arné Clevert, Ana Conesa, Joaquin Dopazo, Mario Fasold, Sepp Hochreiter, Huixiao Hong, Nadereh Jafari, David P. Kreil, Paweł P. Łabaj, Sheng Li, Yang Liao, Simon M. Lin, Joseph Meehan, Christopher E. Mason, Javier Santoyo-Lopez, Robert A. Setterquist, Leming Shi, Wei Shi, Gordon K. Smyth, Nancy Stralis-Pavese, Zhenqiang Su, Weida Tong, Charles Wang, Jian Wang, Joshua Xu, Zhan Ye, Yong Yang, Ying Yu & Marc Salit: 'Assessing technical performance in differential gene expression experiments with external spike-in RNA control ratio mixtures, Nature Communications, 5:5125.
New Publication in Nature Biotechnology
Posted on September 10, 2014

Su Z, Labaj PP, Li S, Thierry-Mieg J, Thierry-Mieg D, Shi W, Wang C, Schroth GP, Setterquist RA, Thompson JF, Jones WD, Xiao W, Xu W, Jensen RV, Kelly R, Xu J, Conesa A, Furlanello C, Gao H, Hong H, Jafari N, Letovsky S, Liao Y, Lu F, Oakeley EJ, Peng Z, Praul CA, Santoyo-Lopez J, Scherer A, Shi T, Smyth GK, Staedtler F, Sykacek P, Tan XX, Thompson EA, Vandesompele J, Wang MD, Wang J, Wolfinger RD, Zavadil J, Auerbach SS, Bao W, Binder H, Blomquist T, Brilliant MH, Bushel PR, Cai W, Catalano JG, Chang CW, Chen T, Chen G, Chen R, Chierici M, Chu TM, Clevert DA, Deng Y, Derti A, Devanarayan V, Dong Z, Dopazo J, Du T, Fang H, Fang Y, Fasold M, Fernandez A, Fischer M, Furió-Tari P, Fuscoe JC, Caimet F, Gaj S, Gandara J, Gao H, Ge W, Gondo Y, Gong B, Gong M, Gong Z, Green B, Guo C, Guo L, Guo LW, Hadfield J, Hellemans J, Hochreiter S, Jia M, Jian M, Johnson CD, Kay S, Kleinjans J, Lababidi S, Levy S, Li QZ, Li L, Li L, Li P, Li Y, Li H, Li J, Li S, Lin SM, López FJ, Lu X, Luo H, Ma X, Meehan J, Megherbi DB, Mei N, Mu B, Ning B, Pandey A, Pérez-Florido J, Perkins RG, Peters R, Phan JH, Pirooznia M, Qian F, Qing T, Rainbow L, Rocca-Serra P, Sambourg L, Sansone SA, Schwartz S, Shah R, Shen J, Smith TM, Stegle O, Stralis-Pavese N, Stupka E, Suzuki Y, Szkotnicki LT, Tinning M, Tu B, van Delft J, Vela-Boza A, Venturini E, Walker SJ, Wan L, Wang W, Wang J, Wang J, Wieben ED, Willey JC, Wu PY, Xuan J, Yang Y, Ye Z, Yin Y, Yu Y, Yuan YC, Zhang J, Zhang KK, Zhang W, Zhang W, Zhang Y, Zhao C, Zheng Y, Zhou Y, Zumbo P, Tong W, Kreil DP, Mason CE, Shi L.: 'A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium', Nature Biotechnology, 32(9):903-914.
Paid internship: Design and development of a Web-App for Next-Generation Sequencing
Posted on July 30, 2014
We are looking for a student with background in web development. The project can be undertaken as part of an internship, as student employee or as part of a bachelor/master/diploma thesis. This project is a collaboration with the bioinformatics group of Prof. Peter F. Stadler at the University of Leipzig.
Find more information in our job flyer (only in German).
New member of the Scientific Advisory Board:
Prof. Dr. Peter F. Stadler
Posted on May 09, 2014

We are very pleased to announce that Prof. Dr. Peter F. Stadler has joined our Scientific Advisory Board.
Prof. Dr. Peter F. Stadler received his Ph.D. in Chemistry from the University of Vienna in 1990 following studies in chemistry, mathematics, physics and astronomy. After a PostDoc at the Max Planck Institute for Biophysical Chemistry in Goettingen he returned to Vienna to work in the area to theoretical biochemistry. Since 1994 he is External Professor at the Santa Fe Institute, a research center focussed on Complex Systems. In 2002 he moved to the University of Leipzig as Full Professor of Bioinformatics. Furthermore, he is Senior Scientific Advisor at the RNomics Group at the Fraunhofer Institute for Cell Therapy and Immunology (IZI) in Leipzig, Principal Investigator at the Center for non-coding RNA in Technology and Health (RTH) in Copenhagen, External Scientific Member of the Max Planck Society affiliated with the MPI for Mathematics in the Sciences in Leipzig, Guest Lecturer at the Institute for Theoretical Chemistry of the University of Vienna and Corresponding Member Abroad of the Austrian Academy of Sciences. He published more than 400 scientific papers and articles and is Member of the Editorial Boards of seven scientific journals.
Celebrating two years ecSeq Bioinformatics
Posted on April 07, 2014
We're celebrating the 2nd Anniversary of ecSeq Bioinformatics and want to thank all clients and workshop participants for the awesome two years. You placed confidence and trust in us to provide a needed service - we did not take this responsibility lightly. We would like to express gratitude to our partners and our great team.
Announcing the 2014 NGS workshops
Posted on March 28, 2014
Do you think your skills in sequencing data analysis could need some improvement? Then our NGS workshop series taking place in October 2014 might be interesting for you. As usual, these hands-on workshops have a strong focus on practical experience meaning that you analyze typical NGS datasets. Our trainers will directly interact with you and give valuable feedback.
Our workshop series 2014 will consist of four separate courses:
For beginners, we have the RNA-seq Bioinformatics: A Practical Introduction course which will cover all the basics required for NGS analysis like file formats, quality control, read processing and read alignment, all based on an example RNA-seq dataset.
We are very happy that we could convince Dr. Michael Hackenberg, the developer of one of the most-used microRNA gene prediction tools for small RNA-seq data (miRanalyzer) to join us for our microRNA workshop. This course titled microRNA Analysis Using High-Throughput Sequencing will cover the essential steps of small RNA-seq analysis from microRNA prediction to extensive downstream analyses.
We also have a workshop on DNA Methylation Analysi where Helene Kretzschmer will show you how she took care of her data in the MMML-Seq study of the International Cancer Genome Consortium (ICGC).
Lastly, there is the workshop Discovering standard and non-standard RNA transcripts where Gero Doose will train you on advanced methods of transcriptome analysis. Gero has a strong background in transcriptome analysis, finding different isoforms, fusion-transcripts or circularized RNAs.
small RNA-Seq Presentation at Forschungsmuseum KOENIG
Posted on March 10, 2014
At March 27, the presentation 'Inferring non-coding RNAs from RNA-Seq Data' will be held in Bonn, Germany.
NGS Workshop at 3rd Programming for Evolutionary Biology Course
Posted on March 05, 2014
At March 22, the presentation 'Analysis of high-throughput sequencing data' will be given at the 3rd Programming for Evolutionary Biology Course in Leipzig, Germany.
New Publication in Genome Biology
Posted on February 10, 2014
Hoffmann S, Otto C, Doose G, Tanzer A, Langenberger D, Christ S, Kunz M, Holdt L, Teupser D, Hackermüeller J, Stadler PF: 'A multi-split mapping algorithm for circular RNA, splicing, trans-splicing, and fusion detection', Genome Biology, 15:R34, doi:10.1186/gb-2014-15-2-r34
New Book Chapter in Methods in Molecular Biology
Posted on January 28, 2014
Hertel J, Langenberger D, Stadler PF: 'Computational prediction of microRNA genes' in J. Gorodkin, W.L. Ruzzo, (eds).: RNA Sequence, Structure and Function: Computational and Bioinformatic Methods. Volume 1097 of Methods in Molecular Biology, Humana Press, New York.
Poster Presentation at 55th ASH Annual Meeting and Exposition
Posted on December 05, 2013
Hezaveh K, Bernhart S, Hoffmann S, Langenberger D, Schlesner M, Stadler PF, Binder V, Lenze D, Siebert R, Hummel M, Borkhardt A: 'In-Depth miRNA Profiling Of Germinal Center Derived B-Cell Lymphomas By Next Generation Sequencing: A Report From The German Icgc-Mmml-Seq Project', Blood 122 (21), 2500-2500.
New Publication in Nature Magazine
Posted on August 16, 2013

Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behiati S, Biankin AV, Bignell GR, Bolli N, Borg A, Boerresen-Dale A, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjord JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinsk M, Jäger N, Jones DTS, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt ANJ, Valdés-Mas R, van Buuren MM, van 't Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Gen Initiative, ICGC Breast Cancer Consortium, ICGC MMML-Seq Consortium, ICGC PedBrain, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR: 'Signatures of mutational processes in human cancer', Nature, 500(7463):415-21.